
La genética traza el mapa de la dispersión mundial del virus
Los genetistas y biólogos computacionales tratan de dibujar el árbol genealógico de las mutaciones del SARS-CoV-2 a medida que se dispersa. El análisis de más de 500 genomas de los cinco continentes no muestra cambios en su virulencia por ahora, ni la aparición de linajes distintos.
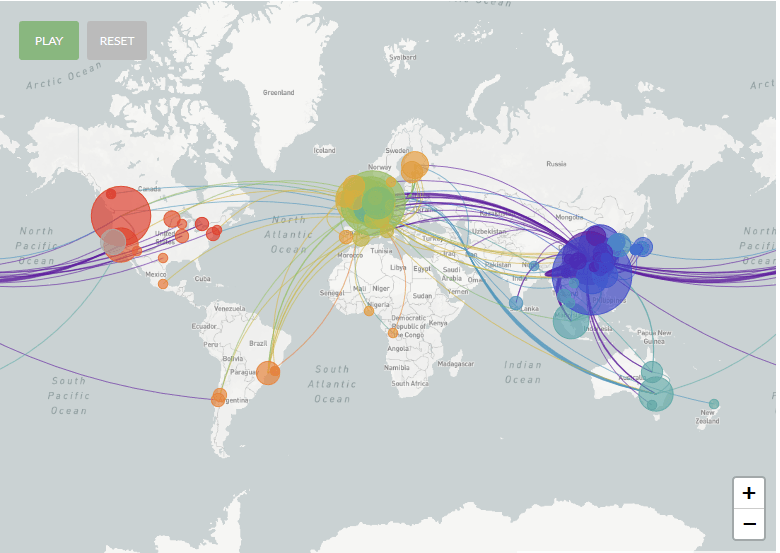
Mapa global de Nextstrain con la reconstrucción de la propagación geográfica de los virus SARS-CoV-2. Se visualizan 529 genomas muestreados entre diciembre de 2019 y marzo de 2020. / Nextstrain
“El COVID-19 circula fluidamente en toda Europa, con mucho movimiento entre países”, dice el último informe del proyecto de código abierto Nextstrain, que pretende aprovechar “el potencial científico y para la salud pública” del análisis del genoma de patógenos.
En el caso del nuevo coronavirus, la comparación de genomas informa de su tráfico entre países y de cómo está mutando, algo esencial para saber si cambia su virulencia. Nada por ahora sugiere que esto esté ocurriendo. Tampoco se observan dos cepas, como se ha especulado en un trabajo ya desacreditado por expertos.
Nextstrain muestra —también gráficamente— el análisis de 512 genomas de pacientes de 33 países en cinco continentes, y la cifra crece día a día. Desde el pasado viernes España ya está en los mapas de Nextstrain de los movimientos del coronavirus, y no —desde luego— porque el virus no estuviera antes aquí, sino porque aún no había ninguna secuencia de pacientes españoles. Las primeras las ha incorporado el grupo de Fernando González, del Servicio de Secuenciación y Bioinformática y el grupo de investigación en Epidemiología Molecular de FISABIO, en Valencia.
El disponer ya de tantas secuencias “es un logro muy importante”, escriben los autores del último informe en Nextstrain sobre el COVID-19, del 13 de marzo. “La secuenciación de un virus desconocido de ARN en medio de una pandemia ha sido posible gracias al trabajo sacrificado y la cooperación para compartir datos de muchos científicos y médicos en todo el mundo”.
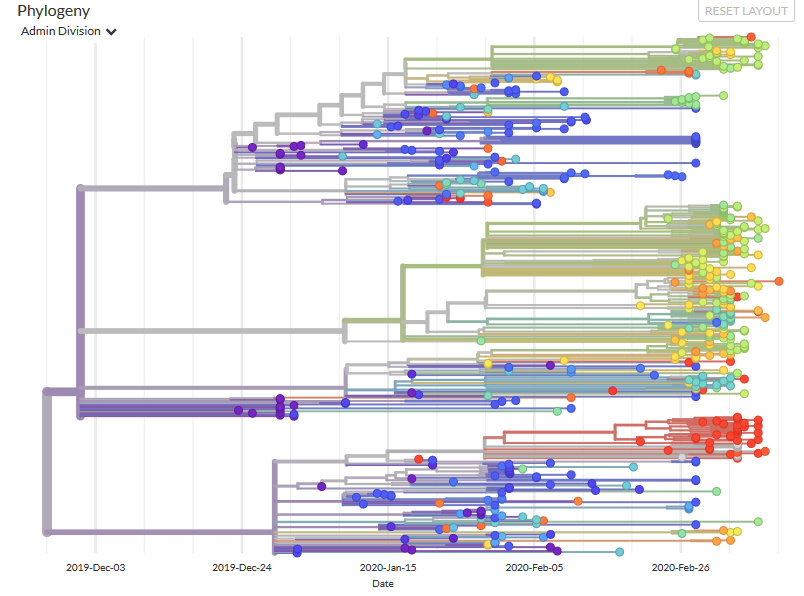
La filogenia muestra las relaciones evolutivas de los virus SARS-CoV-2 de la actual pandemia. Hay una aparición inicial en Wuhan, China, en noviembre y diciembre de 2019, seguida de una transmisión sostenida de persona a persona. Aunque las relaciones genéticas entre los virus muestreados son bastante claras, existe una considerable incertidumbre en cuanto a las estimaciones de las fechas de transmisión y en la reconstrucción de la propagación geográfica. / Nextrain
Los datos actuales “permiten hacer importantes inferencias acerca del brote del virus y monitorear su propagación en tiempo real”, señala el trabajo, cuyo último firmante es Tim Bradford, del instituto Fred Hutch en Seattle, EEUU.
Pero también advierte que “estas conclusiones son limitadas en su representatividad a nivel mundial”. Es decir, que no se hable apenas de lo que ocurre en el hemisferio sur no implica que estén libres de virus sino que aún no hay secuencias de esos países.
No hay una variante más agresiva
Las llamadas a no interpretar los datos al primer golpe de vista no solo atañen al público en general. También los científicos, en su afán por avanzar rápido, llegan a conclusiones apresuradas.
A finales de febrero —cuenta un reciente reportaje en Science—, el propio Bedford comentó en redes sociales que un brote de Italia podría proceder de un paciente alemán, una conclusión que no apoyaban los datos. Las correcciones de expertos llegaron rápidamente, pero lectores no tan expertos ya habían exigido una disculpa a Alemania. Bedford admite en Science que “debí haber sido más cuidadoso con ese hilo de Twitter”.
También fue una mala interpretación de los datos lo que condujo a investigadores chinos a publicar el 3 de marzo un trabajo que concluía que circulaban ya dos variantes del SARS-CoV-2, de las que una era más virulenta. La respuesta de la comunidad fue rápida y dura: “Las conclusiones son claramente infundadas, aumentan el riesgo de difundir desinformación peligrosa en un momento crucial de la epidemia”, escribieron investigadores de la Universidad de Glasgow (Reino Unido).
La realidad es que, aunque más de 500 secuencias parecen muchas, es aún muy poco para sacar conclusiones firmes relativas a la dispersión del nuevo coronavirus. Se necesitan muchas más secuencias, que científicos en todo el mundo se afanan por conseguir.
Fuente: SINC
Derechos: Creative Commons.






















.png)








No hay comentarios:
Publicar un comentario