Una revisión en profundidad: las evidencias actuales | 05 FEB 19
Esclerosis lateral amiotrófica
Conceptos actuales sobfre el proceso patológico, su epidemiología, el estándar de atención y nuevos tratamientos
Autor: Björn Oskarsson,; Tania F. Gendron, Nathan Mayo Clin Proc. 2018;93(11):1617-1628.
Página 1
La esclerosis lateral amiotrófica (ELA) es un trastorno neurodegenerativo mortal que, por definición, afecta las neuronas motoras superiores e inferiores ( en EE. UU., el enfermedad suele denominarse Enfermedad de Lou Gehrig). Además de las neuronas motoras, también pueden estar afectadas las neuronas motoras de la corteza frontal y otras regiones neuroanatómicas.
Destacados del artículo
|
| Antecedentes |
Estas características de la neurona motora inferior contribuyen a la mortalidad más que la pérdida de las neuronas motoras superiores en el cerebro, que provoca espasticidad, torpeza, hiperreflexia y limitación funcional.
Actualmente, se aprecia que también están involucrados los sistemas extramotoras, aunque en diferentes grados. Por ejemplo, algunos pacientes desarrollan pérdida neuronal en la corteza frontotemporal, y como consecuencia, casi la mitad de los pacientes experimentan signos o síntomas cognitivos y conductuales.
El reconocimiento de la participación extramotora en la ELA, a la que anteriormente no se le había prestado atención, ha mejorado la atención clínica y proporciona nuevos conocimientos en la patogenia de la enfermedad.

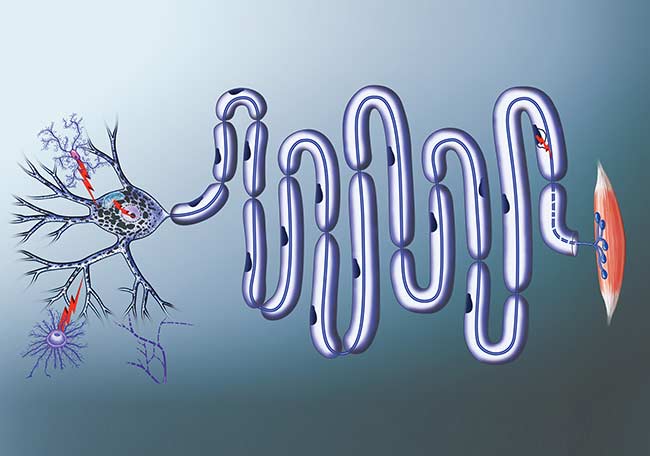
Sistema motor primario que muestra las conexiones entre las neuronas motoras, las superiores e inferiores y el órgano efector muscular. Uper motor neurons: neuronas motoras superiores. Lower motor neurons: nueronas motoras inferiores.
| Epidemiología |
Típicamente, la muerte se produce por fallo ventilatorio. Es 1,5 veces más común en los hombres, y la tasa de progresión de la enfermedad puede ser más rápida en los pacientes con edad avanzada en el inicio, afectación bulbar inicial, deterioro cognitivo y ciertos genotipos.
Respecto de esto último, casi el 10% de los casos es familiar, debido a una mutación genética que generalmente se hereda en forma autosómica dominante, con la participación del hexanucleótido G4C2, y también de diversos genes y proteínas. Las variantes genéticas causan variantes fenotípicas de la enfermedad, con diferencias en su agresividad, edad de inicio, velocidad de progresión y respuesta al tratamiento.
Los avances en el conocimiento de los factores genéticos es muy importante pero el conocimiento de otros factores de riesgo no ha seguido la misma evolución. Dado que las causas genéticas de la ELA se determinan en la concepción y la ELA se desarrolla más a menudo en la edad adulta, posiblemente existan otros factores, como el tiempo y las exposiciones ambientales, que influyen en la susceptibilidad a la enfermedad.
Sin embargo, las relaciones entre los factores de riesgos genéticos y ambientales y el fenotipo de la enfermedad siguen siendo en gran parte desconocidos, posiblemente debido a que los factores de riesgo ambientales son difíciles de identificar, en parte porque la exposición a tales factores puede cambiar con el tiempo, lo que impide que sean registrados con precisión.
Sin embargo, aunque actualmente no se conocen cuáles son los factores de riesgo ambientales que se vinculan irrefutablemente con la ELA, los más sospechosos son: tabaquismo, actividad atlética, servicio militar, ß-N-metilamino-L-alanina, traumatismo craneano, campos electromagnéticos, productos químicos agrícolas, exposición al plomo y otros metales pesados.
En general, el modelo gen-tiempo-ambiente sugiere que el desarrollo de la ELA es un proceso múltiple en el que los defectos genéticos llevan varios pasos hasta que finalmente provocan la ELA.
| Patogénesis |
Una característica común de muchas enfermedades neurodegenerativas es la disfunción neuronal y la eventual muerte celular, que resulta de la acumulación de agregados proteicos en las células de todo el sistema nervioso.
En la mayoría de los casos de ELA, la principal proteína hallada en tales agregados es la denominada TDP-43.Se destaca que las mutaciones en el gen que codifica la TDP-43 han sido descubiertas tanto en la ELA esporádica como en la familiar, lo que refleja un enlace directo entre las anomalías de TDP-43 y la neurodegeneración.
Es probable que este mecanismo implique una combinación de eventos. Las inclusiones TDP-43 pueden ser tóxicas, y también dañar las neuronas mediante el secuestro de TDP-43, evitando que realice sus funciones habituales en las células.
La proteína respuesta transactiva al ADN ligada a la proteína 43 es una proteína de unión al ARN con más de 6.000 dianas de ARN en el cerebro; combinada con su papel en los múltiples pasos del procesamiento del ARN, esto sugiere que la interrupción del metabolismo del ARN contribuye a la patogénesis de la ELA. Este concepto está apoyado por los hallazgos en el comportamiento de diversas proteínas.
Las proteínas están implicadas en la etiología de la ELA. A través de funciones realizadas tanto el núcleo como el citoplasma, estas proteínas ligadas al ARN determinan el destino de los transcritos de ARN, desde su maduración hasta su degradación. Una de esas funciones críticas es el reclutamiento de transcritos de ARN mensajero (ARNm) en gránulos de estrés, formados por el estrés celular.
Esto facilita la supervivencia celular mediante el silenciamiento de los ARNm que no son necesarios para combatir el estrés, al mismo tiempo que permite la traducción óptima del ARNm presente. Los gránulos de estrés no tienen membrana; son organelas citoplasmáticas compuestas por ARNm, factores de transcripción inicial, ribosomas 40S y proteínas ligadas al ARN. Una vez que el evento estresante ha disminuido, los gránulos de estrés se desarman.
Sin embargo, las perturbaciones en TDP-43 y otras proteínas, entre ellas las ligadas al ARN, pueden deteriorar la dinámica adecuada de los gránulos de estrés y por lo tanto evitar una respuesta adecuada al mismo. Cabe mencionar que la localización de TDP-43 y las proteínas ligadas al ARN, relacionadas con los gránulos de estrés, pueden promover aún más la agregación y alteración de la homeostasis neuronal.
Además de los defectos en el metabolismo del ARN alterado, se cree que el metabolismo proteico también contribuye a la patogénesis de la ELA. De hecho, las mutaciones genéticas en la depuración proteica, como la proteína 2B de cuerpos multivesiculares cargada, la optineurina, la secuestosoma 1, la proteína que contiene valosina, la TANK-quinasa de unión 1 y la ubicuitina 2, causan ELA o demencia frontotemporal.
Dos proteínas importantes en las vías de degradación celular son la vía de la ubicuitina proteasoma, que degrada las proteínas solubles de vida corta, y la vía de la autofagia en los lisosomas, que degrada relativamente las proteínas de vida larga, las mal plegadas y agregadas y, las organelas.
Los defectos en estos sistemas de degradación no solo pueden contribuir a la acumulación aberrante de TDP-43 u otras proteínas, sino también complicar aún más el problema, al fallar la eliminación de los gránulos de estrés y, por lo tanto, promoviendo la agregación de esas proteínas. A su vez, se supone que los agregados proteicos inhiben la proteína y secuestran ARN y otras proteínas necesarias para un correcto funcionamiento celular.
Se ha implicado a muchos mecanismos patológicos adicionales en varios pasos del proceso neurodegenerativo de la ELA, como disfunción mitocondrial, daño oxidativo, excitotoxicidad, inflamación y defectos en el transporte intracelular y nucleocitoplasmático.
| La ELA no es solo un trastorno neuronal sino también implica, y posiblemente requiere, otros tipos de células, incluyendo astrocitos, microglía, macrófagos y, potencialmente, oligodendrocitos. |
Finalmente, el concepto de ELA como enfermedad única también ha sido cuestionado. Hay muchas variantes genéticas y, tal vez, los factores de riesgo ambientales que pueden conducir al desarrollo de fenotipos clínicos como la enfermedad de la neurona motora superior, la enfermedad de la neurona motora inferior e, incluso, la demencia frontotemporal, probablemente sean las causas de la ELA, a través de mecanismos patológicos comunes y distintos.
De hecho, se ha especulado que muchos ensayos clínicos de ELA han fallado, al menos en parte, debido a las diferencias potenciales en la fisiopatología subyacente. Se espera que la mayor comprensión de las bases moleculares de las diversas formas de ELA y el desarrollo de modelos preclínicos representativos permitan Identificar los tratamientos efectivos para la ELA. revela las enfermedades que conformarían él diagnóstico diferencial.
Las presentaciones clínicas pueden ir desde un patrón espástico del habla, causado por la pérdida de la neurona motora superior, a una caída del pie causada por la pérdida de la neurona motora inferior, con numerosas variantes en intermedias.
| Enfermedades comúnmente consideradas en el diagnóstico diferencial de la ELA | ||
| Region comprometida | Signos de nueroma motora superior | Signos de neurona motora inferior |
| Bulbar | Lesión de tronco encefálico: (ACV; esclerosis múltiple, tumor | Lesión del tronco encefálico (ACV, esclerosis múltiple, tumor), trastorno de la unión neuromuscular (miastenia gravis, tirosina cinasa muscular específica, miastenia), atrofia bulboespinal uscular. |
| Cervical | Mielopatía cervical | Neuropatía motora multifocal; radiculopatía cervical. |
| Lumbossacra | Mielopatía torácica | Radiculopatía lumbosacera |
| Atención clínica |
Por lo tanto, al comienzo, el diagnostico puede ser incierto y solo puede establecerse con más certeza cuando se desarrollan otras características. Es primordial excluir apropiadamente las otras enfermedades del diagnóstico diferencial. La prueba diagnóstica más relevante es la de base electrofisiológica (electromiografía y estudios de conducción nerviosa).
Otras pruebas diagnósticas de gran valor son las neuroimágenes y los estudios serológicos. El análisis del líquido cefalorraquídeo y las biopsias nerviosas y musculares son pruebas que rara vez se necesitan para excluir enfermedades específicas del diagnóstico diferencial.
Importantes sociedades médicas han desarrollado guías para la práctica, pero existen diferencias provenientes del uso de pruebas y datos nuevos que van surgieron a partir de su respectiva publicación, aunque también hay diferencias metodológicas. Otra diferencia es el alcance de la guía.
La guía del National Institue of Health and Care Excellence, que es más abarcadora, recomienda que antes de las pruebas diagnósticas el paciente sea derivado rápidamente a un neurólogo; posteriormente enfatizan la necesidad de integrar a los prestadores de atención primaria.
Todas las recomendaciones se centran en un enfoque multidisciplinar, lo que ha demostrado ser efectivo, pero, En EE. UU. existe la limitación de que este modelo de atención puede no ser reembolsado en su totalidad. En la actualidad, como un complemento al modelo actual de atención en centros especializados, está surgiendo la telemedicina.
Hasta hace poco, el tratamiento modificador de la enfermedad para la ELA estaba limitado al riluzol, un inhibidor de la liberación de glutamato, aprobado para el tratamiento de la ELA en 1995. El medicamento es generalmente bien tolerado, pero tiene una potencia limitada.
En los ensayos clínicos se ha observado un beneficio de supervivencia de unos meses, sin ningún efecto sobre la función o la calidad de vida discernible. Se han realizado intentos para identificar subgrupos de pacientes más propensos a beneficiarse con el tratamiento con riluzol.
Muchos pacientes utilizan terapias complementarias y alternativas como las vitaminas y los suplementos nutricionales. Algunos suplementos tienen fundamento científico, pero no hay pruebas de ensayos clínicos suficientes para fundamentar su uso.
Se aconseja asesorar al paciente para evitar el uso de tratamientos peligrosos. Con la limitada potencia que poseen los agentes modificadores de la enfermedad, se propone la derivación a un centro para el tratamiento específico de los síntomas
Figura 2

Síntomas tratables • Humor •Energía • Afectación seudobulbar • Alteraciones cognitivas • Comunicación asistida • Deglución • Secreciones • Dolor de hombro • Tastornos ventilatorios torácicos • Constipación • Urgencia urinaria • Funcionalidad de las manos • Motilidad • Calambres |
Esta atención incluye evaluaciones cognitivas y de la conducta, dado que el pensamiento y los síntomas del comportamiento son cada vez más reconocidos en los pacientes con ELA, y pueden afectar el curso de la enfermedad, el manejo de los síntomas y la toma de decisiones durante toda su evolución.
Las intervenciones clínicas son más difíciles en los pacientes con deterioro cognitivo, pero hay estrategias especiales, como la educación adicional y cognitiva recurrente, que puede ayudar a mejorar la adherencia. Lamentablemente, dicen los autores, no hay medicamentos con un beneficio claro para la disfunción cognitiva y conductual, incluyendo los inhibidores de la colinesterasa, utilizados en la demencia amnésica.
Aproximadamente el 25% al 50% de los pacientes con ELA desarrollan efectos pseudobulbares, que se manifiestan como trastorno de expresión emocional inapropiada, caracterizado por una tendencia a llorar o reír sin control o en forma desproporcionada a la emoción experimentada.Estos arrebatos pueden ser incongruentes con el estado de ánimo, ya que la tristeza puede manifestarse como risa, o la alegría como llanto. Existen varios medicamentos efectivos para el control del síntoma, entre ellos una combinación de dextrometorfano y quinidina.
Un diagnóstico de ELA es un estresante psicológico mayor, y aunque la depresión y la ansiedad no son necesariamente más comunes en la población con ELA, en comparación con la población general, es probable que la depresión y la desesperanza sean factores pronósticos negativos en esta enfermedad y, por lo tanto, deben ser tratados cuando están presentes.
El asesoramiento y el apoyo de los grupos de pares para pacientes y cuidadores suelen ser de mucho valor. La fatiga (que empeora los problemas respiratorios) es otro síntoma común que ha sido tratado con éxito con estimulantes. Aunque se piensa que la ELA es indolora, la mayoría de los pacientes experimenta dolor no neuropático que debe ser manejado de acuerdo con su origen.
Los calambres musculares afectan a la mayoría de los pacientes con ELA y pueden ser dolorosos. La mexiletina está emergiendo como un posible tratamiento y como una alternativa a la quinina, que ya no es más recomendada por la FDA (Food and Drug Administration) de EE. UU. para uso clínico. La causa principal de muerte de los pacientes con ELA es la falla ventilatoria.
Las pruebas de función pulmonar, la oximetría de pulso nocturna y los síntomas pueden ayudar a medir la progresión de la debilidad respiratoria.
La ventilación asistida no invasiva, generalmente con presión positiva de la vía aérea, a 2 niveles, puede prolongar sustancialmente la vida y también ayudar a mejorar su calidad. La tendencia en los últimos años, avalada por estudios, ha sido usar el modo volumen controlado en lugar del control de presión, por ser mejor tolerado y más efectivo.
Estos modos de ventilación pueden ser administrados por máquinas de diferente complejidad. A menudo, con el tiempo, la ventilación no invasiva suele fallar, lo que requiere una decisión sobre proceder con una interfaz invasiva (por ej., traqueotomía). En EE. UU. esta opción solo se utiliza en una minoría de pacientes con ELA. En esta decisión, las preferencias del médico y el cuidador probablemente son factores influyentes.
| Es importante informar a los médicos y cuidadores que la no implementación oportuna de la traqueotomía puede llevar a una muerte prevenible, y que una traqueotomía de emergencia realizada sin un correcto conocimiento del resultado a largo plazo también puede tener consecuencias negativas. |
Los estimulantes del nervio frénico, también conocidos como marcapasos del diafragma, no son efectivos en la ELA. La pérdida de la función vocal puede ser compensada artificialmente con dispositivos generadores de voz y tableros de escritura menos complejos.
La deglución deficiente puede llevar a la aspiración y la mala nutrición. La ELA conduce a un aumento de las necesidades calóricas, y la pérdida de peso se correlaciona con una supervivencia más corta. En la ELA se observan niveles bajos de vitamina D, pero estudios recientes sugieren que esta vitamina no es un marcador pronóstico independiente.
Las técnicas de deglución segura y las modificaciones de la dieta pueden favorecer una independencia funcional prolongada. La gastrostomía es el tratamiento estándar para asegurar una nutrición adecuada, pero puede o no prolongar la supervivencia y no ser beneficiosa, aparte de proporcionar un modo de administrar alimentos, líquidos y medicamentos. Esta vía de alimentación podría ser necesaria para los pacientes con apoyo ventilatorio continuo, con el fin de Lograr el máximo beneficio del tratamiento ventilatorio.
La sialorrea debida a la ingestión deficiente puede tratarse con anticolinérgicos, inyecciones de toxina botulínica en la glándula salival o, radioterapia. Los aspiradores pueden utilizarse para eliminar secreciones faríngeas por vía oral. Las secreciones espesas pueden ser fluidificadas mediante hidratación, mucolíticos orales o acetilcisteína nebulizada.
Para conservar la independencia funcional se pueden usar muchos dispositivos; algunos de los más usados son los aparatos ortopédicos tobillo-pie, andadores, silla de ruedas, cama ortopédica, equipos de aseo y ascensores. La fisioterapia y la terapia ocupacional así como los programas de ejercicio para retener la función y minimizar el dolor pueden maximizar el beneficio de los equipos y proporcionar un rango de movimiento.
La espasticidad es un síntoma de la neurona motora superior que puede causar dolor y limitar la función pero ocasionalmente proporciona un grado de beneficio funcional al permitir que los pacientes hagan transferencias de pivote u otras actividades donde una extremidad rígida es más útil que la flácida.
El tratamiento principal es el baclofeno y otros relajantes musculares orales, pero también son útiles las inyecciones de toxina botulínica y, con menor frecuencia, el implante de baclofeno.Las bombas también son útiles.
El estreñimiento es un problema frecuente en la ELA, que puede ser manejado mediante aporte de fluidos, fibra y laxantes.
Otro síntoma es la urgencia urinaria, que se puede tratar con anticolinérgicos y toxina botulínica intravesical. No está claro si estos últimos síntomas son puramente secundarios a la inmovilidad o se deben al compromiso del sistema nervioso autonómico por la ELA.
Los objetivos de la atención médica para cada paciente son importantes, y deben adoptarse planes terapéuticos que tengan en cuenta los objetivos y utilicen intervenciones aceptables para el paciente.
La mayoría de los pacientes con ELA morirá por esta enfermedad, y muchos tendrán preguntas sobre este proceso. Si el paciente solicita este tipo de información, la misma debe ser objetiva y compasiva en relación con el proceso de morir, lo que puede ayudar a desmitificar y reducir la ansiedad.
Para la mayoría de los pacientes, la muerte puede ser descrita como pacífica y terminal, mientras que la ansiedad terminal y la disnea pueden ser controladas con oxígeno, opiáceos y benzodiazepinas. Siempre que sea apropiado se debe dar información relacionada con el cuidado en el hospicio y el control de los síntomas terminales.
Los pacientes suelen preguntar sobre el suicidio asistido por un médico, el que ahora es legal en muchos estados de EE. UU. Esto es una cuestión ética y jurídica compleja, pero la aceptación del suicidio activo como una opción apropiada de muerte por debilidad ventilatoria no tratada va en aumento.
La mayoría de los pacientes con ELA no usa esta opción, incluso en los estados donde es legal, y una pequeña minoría de pacientes con ELA decide suicidarse, incluso sin la asistencia de un médico. El deterioro cognitivo acompañado de dificultades para tomar decisiones y tratar problemas complejos pueden hacer que este tipo de discusiones sea aún más difícil.
| Tratamiento aprobado recientemente |
Este medicamento fue aprobado primero en Japón, en 2001, para el tratamiento de los accidentes cerebrovasculares y ya ha sido usado en más de 1,7 millones de pacientes.Es un depurador de radicales libres que más tarde fue evaluado para otros trastornos neurológicos, incluyendo la ELA. Pero aún existen cuestionamientos sobre los resultados, aunque los mismos fueron bastante notables, mostrando una mejoría en los puntajes de progresión de la enfermedad. Las medidas de la calidad de vida de vida y respiratorias también mejoraron pero sin alcanzar un resultado estadísticamente significativo.
La edaravona se administra diariamente por infusión intravenosa durante 2 semanas seguidas de 2 semanas sin administración del fármaco.
Los ciclos de infusión posteriores permiten interrupciones los fines de semana. La vida media del fármaco es de 4,5 a 6,0 horas, sugiriendo que un horario de dosificación más frecuente podría ser más beneficioso. La mayoría de los pacientes de un estudio de edaravona también tomaban riluzol, lo que muestra que ambos medicamentos pueden usarse juntos. Posee un perfil de seguridad favorable.
Las reacciones más graves reportadas son las alérgicas, incluida la anafilaxia. La edaravona no tiene ninguna interacción farmacológica conocida y no es necesario ajustar la dosis en caso de disfunción renal o hepática moderada.
Su costo y la frecuencia de las infusiones ha aumentado aún más el costo de la atención de la ELA en EE. UU. donde muchas empresas de seguros de salud y la Administración de Salud de Veteranos limitan la cobertura del medicamento, de modo que no todos los pacientes con ELA tienen cobertura.
Como parte de su aprobación, la FDA ha solicitado estudios adicionales para evaluar el efecto de una mayor frecuencia y dosis. La edaravona también se está desarrollando en forma oral, lo que evitaría los principales obstáculos para su administración.
| Tratamiento futuros |
| Células madre |
Los enfoques actuales con células madre están diseñados principalmente para ayudar a proteger las neuronas motoras sobrevivientes, a través de efectos paracrinos (neuroprotección); no están diseñados para reemplazar a las neuronas motoras muertas.
Las células estromales mesenquimáticas (CEM) son las células madre utilizadas como autólogas, debido a su capacidad para segregar factores neurotróficos y moduladores del sistema inmunológico, dos mecanismos que atenúan la evolución de la enfermedad en modelos animales.
Las CEM surgen naturalmente de los pericitos de varios tipos de tejidos (grasa, tejido conjuntivo, médula ósea, pulpa dental) y se cree que median la curación del tejido mientras que también podrían diferenciarse en hueso, cartílago y grasa.
Estas células se aíslan de las biopsias de grasa o de médula ósea, se expanden in vitro y luego se inyectan en el espacio tecal, mediante una punción lumbar (con o sin inyecciones intravenosas o intramusculares) o directamente intraespinales.
Algunos grupos también han desarrollado medios condicionados que aumentan la secreción del factor neurotrófico de las CEM, y otros están utilizando estrategias de edición genética para aumentar la secreción de factores específicos.
En general, el perfil de seguridad de las terapias con estas células ha sido bueno, y ante la falta de requerimiento de un tratamiento inmunosupresor concomitante, se las considera beneficiosas. Aunque ninguno de los estudios de fase temprana han sido potenciados para demostrar su eficacia, hubo informes de beneficios subjetivos mientras que los estudios de casos sugieren que existe un subgrupo de pacientes que puede responder a la terapia.
Recientemente, se inició un ensayo clínico en fase 3 sobre la administración intratecal de CEM autólogas que sobreexpresan factores neurotróficos.
Otra estrategia de células madre utiliza precursores de células del linaje neuroglial. Con este enfoque se han desarrollado líneas de células madre del tejido neural fetal, que se inyectan directamente en el cuerno anterior de la médula espinal.
Debido a que las células son alogénicas, se requiere inmunosupresión para prevenir el rechazo del trasplante. Se han completado ensayos clínicos de fase 1 y 2 utilizando células madre obtenidas del tejido espinal del feto humano. Aunque no potenciado por su eficacia, parece haber un beneficio modesto en un subgrupo de pacientes, lo que dio lugar a más investigaciones.
En la actualidad, se están investigando muchos agentes para el tratamiento de la ELA. Uno de ellos es el masitinib, un inhibidor de la tirosinacinasa, pero en Europa todavía no fue aprobado. Los supuestos efectos del fármaco serían similares a los del riluzol o la edaravona, pero faltan más estudios para validar los resultados y ser aprobado.
El conocimiento de que hay variantes genéticas causantes de la ELA proporciona objetivos putativos para la terapia, incluso en ausencia de una comprensión acabada de los mecanismos patológicos iniciados por el gen mutado. La alteración directa de la expresión del gen mutado puede mitigar los pasos críticos en el desarrollo de la enfermedad.
Un ensayo clínico en fase 1, estudió la administración intratecal de oligonucleótidos antisentido, diseñados para inhibir la superóxido dismutasa Cu-Zn, que puede ser útil en los pacientes con ELA familiar SOD1 pero se esperan los resultados de un segundo ensayo. La identificación de genes y proteínas que intervienen en la patogénesis de la ELA también abre un auspicioso panorama para la terapéutica de la ELA
| Conclusión |
Las causas genéticas identificables, que son objetivos farmacológicos importantes, son responsables de una parte de los casos esporádicos y de la mayoría de los casos familiares, pero, existen más casos sin una etiología claramente definida.
Sin embargo, hay avances significativos en cuanto al esclarecimiento de la patogénesis de la ELA, lo que permitirá conocer mejor los factores ambientales que pueden jugar un papel importante.
Recientemente se han aprobado 2 medicamentos, el riluzol y la edaravona, que han demostrado ser eficaces para el tratamiento de la ELA, pero se necesita aumentar el arsenal terapéutico.
Las estrategias de tratamiento actuales son en gran medida paliativas, con el objeto de mejorar la calidad de vida y la supervivencia, cobrando gran importancia la asistencia ventilatoria.
A partir de una mejor comprensión de las causas genéticas subyacentes y de la patogenia, se están en desarrollo muchos nuevos enfoques terapéuticos, que se espera conduzcan a terapias más eficaces.
Resumen y comentario objetivo: Dra. Marta Papponetti
Contenidos relacionados
Los editores le recomiendan continuar con las siguientes lecturas:























.png)








No hay comentarios:
Publicar un comentario