Una revisión imprescindible | 14 ABR 19
Acromegalia
Resumen de las principales características clínicas de la acromegalia y las novedades terapéuticas
Autor: Annamaria Colao, Ludovica F. S. Grasso, Andrea Giustina y colaboradores Nature Reviews Disease Primersvolume 5, Article number: 20
Página 1
| Introducción |
En la mayoría de los casos es inducida por un tumor hipofisario secretor de GH y, más raramente, por la hiperplasia hipofisaria o la secreción ectópica de GH o la liberación de la hormona liberadora de GH (GHRH).
Tanto el gigantismo como la acromegalia son trastornos raros, causados por el exceso de secreción de GH e IGF1, pero, el gigantismo se produce cuando el exceso de GH provoca el crecimiento lineal antes del final de la pubertad y el cierre de las epífisis, mientras que la acromegalia ocurre cuando el exceso de GH se presenta tras el cierre epifisario.
Durante las últimas dos décadas, el mayor conocimiento de la etiología molecular y la genética del gigantismo hipofisario y la acromegalia ha permitido identificar varias causas genéticas, incluyendo las neoplasias endocrinas múltiples (MEN) tipos 1 y 4, el síndrome de McCune–Albright, el complejo de Carey y el adenoma hipofisario familiar aislado. La exposición prolongada al exceso hormonal induce la desfiguración somática progresiva, y a una amplia gama de manifestaciones sistémicas.
| Signos y síntomas principales En el momento del diagnóstico, los pacientes suelen presentar crecimiento excesivo de las extremidades, como el crecimiento exagerado de las manos y los pies; la cara, incluido el prognatismo y la hipertrofia de los tejidos blandos. Otros posibles síntomas son: hiperhidrosis, bocio, artrosis, síndrome del túnel carpiano, fatiga, pólipos colónicos, apnea del sueño, trastornos reproductivos y metabólicos y, enfermedades cardiovasculares, siendo las más comunes la hipertrofia cardíaca, la hipertensión y las arritmias, aunque en raras ocasiones puede haber insuficiencia cardíaca congestiva. |
Por otra parte, en un pequeño subgrupo de pacientes jóvenes con acromegalia se ha Identificado una mutación de la línea germinal en la codificación del gen que codifica la proteína que interactúa con el receptor de hidrocarburos de arilo (AIP) y, en pacientes con síndrome de acrogigantismo pediátrico ligado al cromosoma X (XLAG), siendo uno de los principales descubrimientos en la acromegalia.
Sin embargo, a pesar del mayor conocimiento de la enfermedad, del perfeccionamiento de las herramientas diagnósticas y las nuevas terapias médicas, en un número importante de pacientes la enfermedad no puede ser controlada, incluso cuando se utilizan todos los enfoques terapéuticos disponibles, mientras que los costos económicos, la mortalidad y la morbilidad siguen siendo elevados y la calidad de vida sigue siendo baja.
| Epidemiología |
Antes del año 2000, la prevalencia global era de <7 casos/100,000 individuos. Sin embargo, en los últimos 5 años, el estudios de acromegalia de poblaciones de Islandia y Malta reportaron una prevalencia de >13 casos/100.000 individuos mientras que en Suecia, España y Dinamarca se reportó una prevalencia más baja (3,6–3,9 casos/100.000 individuos): Los autores atribuyen estas diferencias a un sesgo de selección que pudo ocasionar una subestimación de la prevalencia.
No se halló diferente prevalencia entre ambos sexos. La mayor prevalencia en estudios recientes puede estar vinculada a mayor rigurosidad metodológica para el enfoque diagnóstico. También el diagnóstico en estudios de población recientes puede haber mejorado por contar con grandes bases de datos médicas locales, o deberse a una mayor supervivencia, debido al mejoramiento de las técnicas quirúrgicas, el seguimiento postquirúrgico y el tratamiento médico.
> Incidencia
Los autores mencionan varias causas que pudieron haber contribuido a una mayor tasa de diagnóstico: disponibilidad de criterios diagnósticos universalmente aceptados después del año 2000; advenimiento de medios e información sociales y divulgación por páginas web; incremento verdadero de la incidencia de la enfermedad por efecto ambiental, como los cambios vinculados a la intensa contaminación (hipótesis poco investigada). La edad media al hacer el diagnóstico es 40-50 años.
El lapso desde la aparición de los síntomas hasta el diagnóstico ahora es menor (estimado promedio ≤5 años), aunque en algunos casos los intervalos son mucho más largos (hasta 25 años). Esta estimación se basa principalmente en el recuerdo del paciente y no en los registros médicos.
Como la duración de la enfermedad activa es la principal determinante de gravedad de la mayoría de las complicaciones de la acromegalia, incluyendo el mayor volumen del tumor al momento del diagnóstico, estos datos enfatizan la importancia de la educación superior y mayor conciencia de la enfermedad entre los clínicos y los pacientes.
Más del 95% de los pacientes con acromegalia tienen un adenoma secretor de GH esporádico
| Causas de acromegalia La acromegalia es una enfermedad lentamente progresiva que resulta del aumento de la liberación de hormona del crecimiento (GH) y, en consecuencia, del factor de crecimiento símil insulina I (IGF1). El exceso de liberación de GH es inducido en la mayoría de los casos por uncáncer hipofisario secretor de GH y, más raramente, por la hiperplasia hipofisaria o la secreción ectópica de hormona liberadora de GH (GHRH). La acromegalia iatrogénica puede ocurrir con la sobredosis o el reemplazo inadecuado de GH. En raras ocasiones, la acromegalia se asocia con síndromes genéticos, incluyendo la neoplasia endocrina múltiple tipo 1, el síndrome de McCune-Albright, acromegalia familiar y complejo de Carney. |
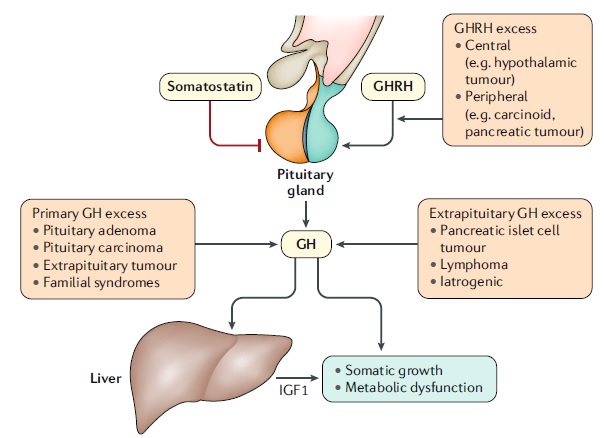
Causas de la acromegalia. La acromegalia es una enfermedad lentamente progresiva que resulta de una mayor liberación de la hormona del crecimiento (GH) y, en consecuencia, del factor de crecimiento tipo insulina I (IGF1). El exceso de liberación de GH es inducido en la mayoría de los casos por un tumor hipofisario secretor de GH y más raramente como resultado de hiperplasia hipofisaria o GH ectópica o secreción de hormona liberadora de GH (GHRH). La acromegalia iatrogénica puede ocurrir con una sobredosis o un reemplazo inadecuado de GH. En raras ocasiones, la acromegalia se asocia con síndromes genéticos, que incluyen neoplasia endocrina múltiple tipo 1, síndrome de McCune-Albright, acromegalia familiar y complejo de Carney.
Para obtener información adicional sobre su prevalencia epidemiológica se han realizado estudios en individuos con síntomas que generalmente se asocian con acromegalia.
Por ejemplo en pacientes con apnea del sueño, llegando a la conclusión de que el cribado de acromegalia en los individuos con apnea del sueño no está indicado, a menos que las características clínicas clásicas estén presentes. Se han publicado casos (48/100.000) ocasionales de acromegalia en pacientes con diabetes mellitus tipo 2.
| Mecanismos y fisiopatología |
Más del 95% de los pacientes con acromegalia tienen un adenoma secretor de GH esporádico que surge de células somatotropas (una célula hipofisaria que secreta GH) o de células de secreción mixta de GH y prolactina, en la hipófisis anterior. En raras ocasiones, la acromegalia proviene de un tumor familiar o de la producción ectópica de GH o GHRH.
Los adenomas secretores de GH se desarrollan como consecuencia de la proliferación desregulada de las células somatotropas altamente diferenciadas que expresan una producción excesiva de GH y, como resultado, de la mayor síntesis y secreción de GH por las células somatotropas. Estos adenomas son invariablemente benignos y no producen metástasis, incluso si son localmente invasivos, pero pueden crecer agresivamente con invasión local y se clasifican según la morfología celular, la expresión del producto génico, la intensidad del granulado intracelular de GH y la invasividad.
Los adenomas tipo 1 son más pequeños, densamente granulados, se desarrollan sobre todo en los pacientes mayores y se asocian con un grado relativamente leve de la enfermedad y, por lo tanto, en su mayoría son los que evolucionan más favorablemente.
Por el contrario, los adenomas tipo 3 son poco granulados, más grandes, más invasivos y se presentan predominantemente en los pacientes más jóvenes. En general, son más resistentes al tratamiento que los adenomas tipo 1 y 2 y tienen comorbilidades adversas. Los adenomas tipo 2 tienen fenotipos intermedios.
La patogénesis de los tumores esporádicos secretores de GH sigue siendo poco conocida, aunque se han observado anomalías en el factor de crecimiento o en la expresión del receptor del factor de crecimiento, desregulación del ciclo celular y de la transducción de señales, alteración de la expresión de genes del ciclo celular y, pérdida de la expresión del gen supresor tumoral.
> Desregulación de la señal de transducción
La interrupción de la señalización de las células tumorales somatotropas incluye la activación de las mutaciones somáticas en el gen GNAS1, que codifica a la subunidad α de la proteína G ligada al nucleótido guanina, que aparecen hasta en el 40% de los adenomas secretores de GH. Estas mutaciones también están presentes en pacientes con el síndrome de McCune-Albright.
La señalización de la somatostatina hipotalámica mediante receptores de superficie de las células somatotropas suprime la producción de GH y la mutación GNAS1, lo que también confiere una respuesta preferencial a los ligandos de los receptores de somatostatina, los cuales son inhibidores farmacológicos de la GH.
La GHRH hipotalámica es un factor mitogénico que induce la producción de cAMP, lo que conduce a una proliferación somatotrofa, además de una mayor secreción de GH, mientras que la reducción de la somatostatina hipotalámica reduce la hipersecreción de GH.
Otro objetivo terapéutico en los tumores somatotróficos podría ser el traductor de señales y activador de transcripción 3 (STAT3), que es otra molécula de señalización que ha sido implicada en la hipersecreción de GH por las células somatotropas. Los tumores secretores de GH, expresan más STAT3, lo que provoca mayor síntesis de GH.
Por otra parte, la abundancia de STAT3 se correlaciona con el grado de hipersecreción de GH mientras que la inhibición de la actividad de STAT3 bloquea el crecimiento de los autoinjertos de tumores somatotróficos e inhibe la secreción de GH por las células cultivas derivadas de tumores somatotróficos humanos.
Como la GH también induce la fosforilación de STAT3 y la translocación de las células somatotróficas, el mecanismo subyacente en la hipersecreción de GH en los adenomas somatotróficos podría ser la retroalimentación autocrina o paracrina positiva para inducir STAT3.
Estudios recientes sugieren la existencia de una respuesta aberrante de las células somatotropas al polipéptido insulinotrópico dependiente de glucosa o a la existencia de un exceso de expresión del receptor del polipéptido insulinotrópico dependiente de glucosa en las células somatotropas, que podría explicar el aumento paradójico de la GH en un subgrupo de pacientes con acromegalia después de la prueba de tolerancia a la glucosa oral, contrarrestando la actividad del inhibidor hipotalámico, inducida por la glucosa.
> Interrupción del ciclo celular
Además de los defectos de señalización, las células secretoras de GH de los tumores hipofisarios suelen caracterizarse por aneuploidía, daño del ADN y alteración del ciclo celular, incluida la detención prematura del ciclo celular.
Por ejemplo, el exceso de expresión del gen transformador de proteína 1 en el tumor hipofisario, el índice de securina de mamíferos que asegura la fidelidad de la separación cromosómica en los tumores secretores de GH, resulta en la interrupción del ciclo celular. Los niveles excesivos de este gen en los adenomas se deben a defectos de la sumoilación de la proteína y se correlacionan con la invasividad tumoral.
En general, las interrupciones del ciclo celular y las restricciones de la proliferación celular, incluyendo la sobreexpresión de inhibidores de la quinasa dependientes de ciclina en particular, la p21 intranuclear conducen a la senescencia celular que actúa como tampón contra la transformación maligna de las células somatotropas. Así, el equilibrio entre las perturbaciones y la inhibición de la proliferación celular que promueven determina si un tumor secretor de GH se torna invasivo.
> Estabilidad genómica
Un estudio de asociación genómica de 128 adenomas secretores de GH identificó 3 loci de susceptibilidad (2 en el cromosoma 10 y 1 en el cromosoma 13) que se asocian con adenomas hipofisarios esporádicos. También se hallaron variaciones en el número de copias.
Consistente con defectos del ciclo celular o de la señalización, la secuenciación del genoma completo y el análisis del número de copias somáticas no detectaron mutaciones oncogénicas clásicas en los adenomas de células somatotropas.
Por otra parte, el perfil genómico de los tumores secretores de GH mostró una alteración heterogénea en el número de copias, coincidente con un estado de inestabilidad genómica. El 45% de un subgrupo de tumores que también contenían mutaciones del gen GNAS1 se observó alteración genómica y aneuploidía.
> Expresión alterada de GHRH o GH
La secreción ectópica de GHRH se produce principalmente a partir de tumores carcinoides de origen pancreático o pulmonar y de gangliocitomas hipotalámicos. El exceso de acción de GHRH puede provocar la hiperplasia o el agrandamiento de la hipófisis y, si los niveles circulantes de GHRH son elevados, surge la indicación de imágenes abdominales o torácicas.
Aunque es extremadamente raro, la producción ectópica de GH puede ocurrir a partir de adenomas del tracto faríngeo o de tumores extrahipofisarios (tumores pancreáticos, linfoma).
> Causas genéticas
En los tumores somatotrofos de grupos con acromegalia familiar y algunas formas de gigantismo, el análisis genómico ha identificado nuevas mutaciones en la línea germinal y mutaciones somáticas que perturban las vías intracelulares.
Por ejemplo, el acrogigantismo ligado al síndrome X (que no es familiar) es un síndrome de gigantismo de inicio muy temprano que se caracteriza por la rápida aceleración del crecimiento a partir de la edad media de 1 año y se asocia con un adenoma hipofisario diferente o con hipersecreción de GH asociada a hiperplasia hipofisaria.
La acromegalia hereditaria es rara y puede estar asociada con el síndrome de neoplasia endocrina múltiple tipo 1 (MEN1) y el complejo de Carney, o puede ocurrir como adenoma hipofisario aislado.
La acromegalia familiar con inicio de la hipersecreción de GH a una edad más temprana, predominantemente debida a un macroadenoma, se asocia con mutaciones en la línea germinal de AIP. En raras ocasiones, estas mutaciones están asociadas a prolactinomas.
Las mutaciones AIP están presentes hasta en el 30% de los pacientes con acromegalia familiar. En <5% de los pacientes con acromegalia esporádica se ha detectado una baja penetrancia de las mutaciones AIP, con predisposición autosómica dominante.
Los polimorfismos AIP son heterogéneos y, cuando están presentes, las mutaciones en AIP no parecen conferir características clínicas particulares en los pacientes con acromegalia esporádica,. Se requieren otros estudio para determinar la importancia de estas mutaciones de AIP en los pacientes adultos con acromegalia esporádica.
| Diagnóstico, cribado y prevención |
Las manifestaciones más frecuentes de la acromegalia son la dismorfia facial y acral, el agrandamiento de manos y pies; dedos de manos y pies rollizos y engrosamiento del tejido blando.Los pacientes tienen un aspecto característico cara rectangular, nariz agrandada y ensanchada, pómulos prominentes, frente abultada, labios engrosados y marcadas arrugas cutáneas. Tienen tendencia al prognatismo, ensanchamiento maxilar, separación de los dientes y maloclusión mandibular.
Las formas graves, crónicas, progresivas, se caracterizan por deformidades esqueléticas, que conducen a la cifosis dorsal y la distorsión de la caja torácica. Sin embargo, actualmente es raro hallar estos signos, debido al diagnóstico precoz. En los pacientes no diagnosticados puede ocurrir una transformación facial lenta, insidiosa, a lo largo de varios años.
Como resultado, cuando un individuo solicita atención médica (más comúnmente debido al crecimiento o hinchazón de las extremidades y deformación de la cara), a menudo es un médico no familiarizado con el paciente (médico general o endocrinólogo) que propone el diagnóstico.
Sin embargo, la mayoría de los pacientes (principalmente mujeres) se quejan de un retraso importante en el diagnóstico, a pesar de los síntomas evidentes y de haber realizado varias consultas médicas.
Cambios físicos • Prominencia de la frente • Prognatismo • Macroglosia • Hiperhidrosia • hipertrofia de tejidos blandos • Agrandamiento de la nariz y los labios |
Complicaciones metabólicas y endócrinas • Tolerancia a ka glucosa alterada • Diabetes mellitus • Resistencia a la insulina • Dislipidemia • Bocio tiroideo • Crecimiento excesivo de las extremidades |
Complicaciones gastrointestinales • Pólipos colónicos • Dolicomegacolon |
> Síntomas inespecíficos y comorbilidades
Además del agrandamiento de las extremidades y facial característico, la acromegalia se asocia con síntomas inespecíficos, como la cefalea (independientemente del tamaño del adenoma hipofisario, siendo ésta la segunda queja inicial que suele conducir al diagnóstico de acromegalia; astenia; hiperhidrosis, especialmente nocturna; acroparestesias (generalmente el síndrome del túnel carpiano) y, artralgias. En las mujeres, también son frecuentes los trastornos menstruales, una razón frecuente de consulta médica.
En el momento del diagnóstico, los pacientes pueden presentar comorbilidades que están relacionadas con los niveles excesivos de GH o del (IGF1), como diabetes o intolerancia a la glucosa, hipertensión, síndrome de apnea del sueño, miocardiopatía (principalmente hipertrofia ventricular izquierda) y bocio. Las artralgias, que tienen un origen mecánico, degenerativo y no inflamatorio, ocurren en el 30 al 70% de los pacientes.
Aunque pueden verse afectadas todas las articulaciones, las más susceptibles son las articulaciones grandes. La prevalencia estimada de la afectación espinal es ~40–50%. Puede haber dolor de espaldas, más frecuente a nivel lumbar. A veces, la claudicación intermitente bilateral puede indicar estenosis espinal lumbar. El dolor también puede estar relacionado con una mayor prevalencia de fracturas vertebrales, a pesar de la normalidad de la densidad mineral ósea.
El examen radiológico tiene características típicas, como la osificación de la superficies anterior y lateral de los cuerpos vertebrales, así como un aspecto bicóncavo y festoneado de los mismos. El síndrome del túnel carpiano sintomático y la neuropatía del nervio cubital en el túnel son frecuentes y lo más probable es que implique el edema del nervio mediano más que la compresión extrínseca, que puede ser fácilmente evaluada por ecografía.
El 20 al 50% de los pacientes sufre hipertensión arterialMuchos presentan miocardiopatía específica sin afectación coronaria (actualmente, la coronariopatía está presente solo en una minoría de pacientes), hipertensión, diabetes y trastornos valvulares. La afectación cardíaca es inicialmente asintomática, y suele consistir en hipertrofia miocárdica. Si los trastornos cardíacos progresan se puede presentar insuficiencia cardíaca congestiva, aunque hoy en día estas complicaciones son mucho menos frecuentes.
- El exceso de GH provoca resistencia a la insulina en el hígado o en la periferia, dando lugar a hiperinsulinemia.
- La prevalencia de diabetes (20–56%) e intolerancia a la glucosa (16–46%) es bastante elevada y depende de la serie de casos.
- Siempre que la menor sensibilidad a la insulina esté compensada por el aumento de la secreción de insulina por las células β pancreáticas, la tolerancia a la glucosa permanece normal.
La apnea del sueño afecta al 60–80% de los pacientes con acromegalia en el momento del diagnóstico y es más frecuente en los hombres. Más comúnmente, la apnea del sueño se investiga en presencia de ronquidos (informado por el 78% de los pacientes con acromegalia) y en aquellos con somnolencia diurna (51%) o fatiga y cefalea matutinas (16%).
Esta apnea suele ser obstructiva, aunque un tercio de los pacientes tiene apnea central. La apnea obstructiva está vinculada a los cambios anatómicos que resultan del crecimiento maxilar y mandibular, el engrosamiento de los tejidos blandos (especialmente paladar y úvula) y los cambios en los ángulos de los diferentes segmentos óseos, que hacen más colapsables las paredes posteriores y laterales de la hipofaringe. La hipertrofia de la lengua y las glándulas submaxilares también tiene un papel en la apnea obstructiva.
Estudios prospectivos han hallado que hasta el 45% de los pacientes con acromegalia tienen pólipos colónicos y, en el 24% de ellos son adenomatosos, pudiendo surgir en cualquier parte del colon.
| Diagnóstico bioquímico de la acromegalia |
De hecho, en los individuos sanos, la hiperglucemia suprime la secreción hipofisaria de GH y, por lo tanto, los niveles séricos de GH disminuyen. Aunque el mecanismo de esta reducción no es del todo conocido, la explicación más plausible parece ser el aumento de la somatostatina hipotalámica mediado por la glucosa.
Cuando un adenoma hipofisario secreta GH de forma autónoma y en exceso, esta regulación fisiológica se pierde y los niveles de GH ya no son suprimidos por la glucosa oral.
> Medición de los niveles séricos del IGF1
La prueba diagnóstica bioquímica de primera línea es la determinación de los niveles de IGF1 y es la recomendada para los pacientes con manifestaciones clínicas de acromegalia típica.De hecho, la medición del IGF1 es más simple que la medición de la GH porque solo requiere una muestra de sangre de cualquier momento del día.
En pacientes sin manifestaciones típicas de acromegalia pero con afecciones asociadas, incluyendo apnea del sueño, diabetes mellitus tipo 2, artritis debilitante, síndrome del túnel carpiano, hiperhidrosis e hipertensión, o que tengan un tumor hipofisario, también se recomienda la determinación del IGF1.
Debido a su importancia clínica en el diagnóstico y seguimiento de la acromegalia, lo autores enfatizan el valor de algunos puntos metodológicos en cuanto a la medición del IGF1.
Se destaca que se hallan concentraciones séricas de IGF1 por encima de la media en el embarazo normal, la pubertad y el período pospuberal, mientras que los niveles por debajo de la media se observan en la diabetes no controlada o la insuficiencia renal. En consecuencia, estos escenarios pueden dificultar el diagnóstico de acromegalia mediante el análisis del IGF1.
> Medición de los niveles de GH
La medición de los niveles de GH es considerada el diagnóstico bioquímico de segunda línea, ya que para hacer el diagnóstico una muestra no suele ser suficiente.
El nivel basal de GH sérica (matinal o al azar)está elevado en la acromegalia. Sin embargo, también se pueden encontrar niveles elevados de GH en individuos sanos debido a la naturaleza episódica de la secreción fisiológica de GH, lo que resulta en niveles de GH que oscilan entre indetectables (la mayoría de las veces) y máximos, pudiendo alcanzar los 30 μg/l.
De hecho, la secreción de GH por la hipófisis está sujeta a la doble regulación de la somatostatina hipotalámica y la GHRH. Cuando predomina la somatostatina (la mayoría de las veces), los niveles de GH son muy bajos, mientras que cuando predomina la GHRH, se observan niveles pico de GH (aproximadamente una docena de picos/día, principalmente durante el sueño). Por lo tanto, la guía clínica de la Endocrine Society recomienda no confiar en las mediciones de los niveles de GH de muestras aleatorias para hacer el diagnóstico de acromegalia.
Por el contrario, en pacientes con niveles séricos elevados o no concluyentes de IGF1, se recomienda confirmar el diagnóstico cuando no se produce la supresión de los niveles de GH (hasta <1 μg/l) después de la hiperglucemia confirmada durante una prueba de tolerancia a la glucosa oral.
Este punto de corte de GH, 1 μg/l, probablemente debería reducirse a ≤0,3 μg/l porque, con el uso generalizado de la quimioluminiscencia sensible o ensayos fluorométricos con límites de detección muy bajos (0,10–0,30 μg/l), algunos pacientes con signos clínicos claros de acromegalia y niveles elevados del IGF1podrían tener niveles de GH suprimidos (<1 μg/l durante la prueba de tolerancia a la glucosa oral) debido a un leve exceso de secreción de la GH.
Sin embargo, acotan los autores, es probable que sea poco realista proponer un punto de corte de GH para esa prueba, ya que los niveles de GH después de la supresión de la glucosa son altamente dependientes del ensayo que se utiliza para medir los niveles de GH.
Sin embargo, los niveles de GH basales y posteriores en la prueba de tolerancia a la glucosa están claramente elevados en la mayoría de los pacientes con acromegalia, y el diagnóstico bioquímico de la enfermedad suele ser sencillo hasta en el 30% de los pacientes, mientras que los niveles de GH aumentan paradójicamente en respuesta a la glucosa oral.
> Análisis genético
En pacientes acromegálicos jóvenes (<30 años) (o con gigantismo, debe hacerse el análisis genético para detectar el gen de AIP. En los niños que experimentan un crecimiento rápido a una edad tan temprana como los 2 a 3 meses, se debe sospechar el acrogigantismo ligado a X, y detectar las microduplicaciones del cromosoma Xq26.3.
El análisis genético de AIP también está indicado cuando en la familia existen casos de acromegalia o adenoma hipofisario (adenoma hipofisario familiar aislado).
Si la acromegalia está asociada con características de MEN1, como el hiperparatiroidismo primario, los tumores neuroendocrinos pancreáticos y los tumores suprarrenales, tanto en el paciente como su familia, se debe proponer el análisis genético del MEN1. Si el análisis genético es positivo en el paciente, se deben investigar las mutaciones en los genes AIP y MEN1 en los familiares.
| Diagnóstico diferencial |
Esta presentación a menudo se acompaña de cefalea repentina muy intensa, que a veces se asocia con parálisis del nervio oculomotor, visible mediante imágenes de TC o resonancia magnética (RM). Sin embargo, a veces se produce la necrosis espontánea, pudiendo llevar a la curación clínicamente silenciosa del adenoma.
La paquidermoperiostosis (o artrosis hipertrófica primaria), un trastorno genético extremadamente raro que conduce a un aumento del nivel de prostaglandina E2, pudiendo ser mal diagnosticado como acromegalia. Sin embargo, en estos pacientes se produce mayor engrosamiento de la piel que en aquellos con acromegalia; dedos en palillo de tambor y periostosis de los huesos tubulares, característicos de esa enfermedad.
En algunos pacientes con deficiencia del miembro 1 de la superfamilia de inmunoglobulinas (IGSF1) pueden hallarse niveles aumentado del IGF1, una causa muy rara de hipotiroidismo central y macroorquidia, que también resulta en mayor circunferencia de la cabeza y la cintura.
Algunos pacientes con resistencia grave a la insulina pueden tener características acromegálicas, a pesar de los niveles normales de GH e IGF1, pero suelen presentarse con otros signos de resistencia a la insulina (acantosis nigricans, hiperinsulinemia) o aspecto lipodistrófico.
Por otra parte, en estas condiciones, la RM hipofisaria es normal, al igual que el tamaño y la morfología de la silla turca, mientras que en los pacientes con acromegalia con resolución espontánea del adenoma, la deformación de la silla turca suele persistir.
| Resonancia magnética |
Los microadenomas (<10 mm de diámetro; 20% de los casos) se ven como Imágenes redondeadas homogéneas, bien circunscritas y, en general, ligeramente hipointensas en secciones ponderadas en T1.
Después de la inyección de gadolinio aparecen hipointensas. Los macroadenomas (>10 mm de diámetro) suelen ser isointensos en secciones ponderadas en T1 sin contraste; el contraste con gadolinio los muestra hiperintensos.
La RM permite el análisis de la expansión extraselar: supraselar, hacia la cisterna optoquiasmática y el quiasma; más abajo, hacia el seno esfenoidal o, lateral, hacia el seno cavernoso. Esta información es importante para el neurocirujano.
La invasión del seno cavernoso es difícil de confirmar porque puede ser que parezca que el adenoma invade el seno cavernoso y solo tratarse de la compresión de la pared medial. Un signo inequívoco de la invasión del seno cavernoso es comprobar que el adenoma reviste por completo la arteria carótida interna intracavernosa. Las diferentes secciones de la RM y el aspecto antes y después de la inyección de gadolinio ayudan a determinar el momento de la cirugía.
De hecho, en el caso del macroadenoma con extensión superior que alcanza el quiasma óptico, la evaluación oftalmológica (agudeza visual y campo visual) es obligatoria para el diagnóstico de compresión del quiasma óptico, que indica la necesidad de la extirpación quirúrgica urgente del tumor. La invasión de estructuras vecinas (en particular, el seno cavernoso) indica la extirpación quirúrgica incompleta del tumor y la necesidad de un tratamiento médico posoperatorio.
La clasificación de adenomas de Knosp en 4 grados indica la probabilidad de la reducción tumoral macroscópica y la remisión hormonal.
Si la RM no revela un adenoma o si la hipófisis se ve asimétrica, abultada, hiperplásica, entonces se debe sospechar la acromegalia secundaria a la secreción ectópica de GHRH.

RM hipofisaria en un paciente con acromegalia que muestra un macroadenoma hipofisario. Secuencias ponderadas en T1, secciones coronal (parte a) y sagital (parte b), después de la inyección de gadolinio que muestran una señal hipointensa en comparación con la pituitaria normal (no mostrada).
Para determinar si las funciones de la hipófisis son deficientes o hay un exceso de secreción es necesario evaluar otras funciones. La función tirotrófica se evalúa midiendo la tiroxina (T4) libre y la triyodotironina (T3) libre y la hormona estimulante de la tiroides (TSH).
La evaluación de la función corticotrófica requiere la medición del cortisol sérico matinal, aunque cuando esta medición no es concluyente, suelen requerirse pruebas de estimulación (prueba de estimulación de la hormona adrenocorticotrófica [ACTH] o prueba de tolerancia a la insulina).
Los ciclos menstruales normales en las mujeres en edad fértil reflejan una función gonadotrófica normal. En los varones, la función gonadotrófica se evalúa midiendo los niveles séricos de testosterona. La hipersecreción de prolactina ocurre en el 30% de los pacientes y puede ser funcional, secundaria a la compresión del tallo hipofisario por el tumor o, a un adenoma mixto.
> Cribado
Como la acromegalia es rara, es deseable reducir el largo retraso hasta establecer el diagnóstico. Algunos investigadores han intentado detectar la acromegalia midiendo los niveles de IGF1 en pacientes con comorbilidades comunes en la acromegalia (por ej., diabetes, síndrome de apnea del sueño o hipertensión).
Sin embargo, dada la muy baja prevalencia de la enfermedad, la detección sistemática mediante esta medición, incluso en poblaciones seleccionadas, no es rentable. Para los autores, es curioso que la acromegalia pueda ser detectada por un programa informático rue utiliza fotografías de la cara, en particular los cambios progresivos en la apariencia facial de un paciente con acromegalia.

| Cambios progresivos en la apariencia facial en una paciente con acromegalia. Sobre la base de un análisis de estas fotografías (y otras que no se muestran), así como el hecho de que el paciente tenía un anillo (comprado en 1988) que no pudo seguir usando en 1990, la enfermedad probablemente comenzó entre 1988 y 1990, 22 años antes del diagnóstico de acromegalia que se hizo en 2011. Adaptado con permiso de la ref.78, Elsevier. |
| Tratamiento |
Con el tiempo, las guías internacionales y declaraciones de consenso han cambiado sustancialmente, debido al mejor conocimiento de la enfermedad, mejores ensayos de GH e IGF1, y disponibilidad de nuevas opciones terapéuticas, logrando un progreso hacia el tratamiento cada vez más personalizado. Este tratamiento permite optimizar la terapia, al considerar las características del paciente, la evolución de la enfermedad y la respuesta a los tratamientos específicos, logrando la reducción de los costos de atención.
Por otro lado, la clasificación de la OMS de los tumores hipofisarios reconoce el papel de la inmunohistoquímica hormonal y otros marcadores inmunohistoquímicos en la mejoría de la clasificación de los adenomas hipofisarios agresivos, permitiendo así que los médicos traten mejor a sus pacientes. En particular, en la clasificación revisada no se recomienda usar el término adenoma atípico.
Además de los subtipos de tumores, es muy recomendable evaluar el potencial proliferativo del tumor (por recuento mitótico) y el índice Ki67) y otros parámetros clínicos, como la invasividad tumoral.
Al evaluar clínicamente los adenomas agresivos, la nueva clasificación también reconoce algunos subtipos de tumores hipofisarios neuroendocrinos, como el adenoma somatotrofo escasamente granulado, ya que son adenomas hipofisarios de alto riesgo por su comportamiento clínico agresivo.
El tratamiento óptimo de los pacientes con acromegalia requiere un equipo multidisciplinario (endocrinólogo, neurocirujano experto en cirugía transesfenoidal, neurorradiólogo y radioterapeuta) experto en tumores hipofisarios y paraselares, y un neuropatólogo especializado en la evaluación histopatológica, incluyendo el análisis de las hormonas hipofisarias y los marcadores proliferativos.
> Normalización de los niveles de IGF1 y GH
En las guías de práctica clínica actuales de la Endocrine Society, el objetivo principal es la normalización de los niveles de GH y IGF1, evaluados con métodos más modernos, inmunoensayos sensibles que se utilizan para el diagnóstico. El objetivo terapéutico es alcanzar un nivel de GH <1,0 μg/l en una muestra al azar y un nivel normal de IGF1.
Un punto de corte de GH <1 μg/l después de la prueba de tolerancia a la glucosa se asocia con mejores resultados a largo plazo y menor riesgo de mortalidad en pacientes que se han sometido a una cirugía, aunque con los ensayos de GH ultrasensibles, las guías más recientes sugieren un punto de corte de GH de 0,4 μg/l.
Sin embargo, la normalización de los niveles de IGF1 es un objetivo clave del tratamiento de la acromegalia, ya que es el mejor predictor de control de la enfermedad. Se recomienda hacer el seguimiento del paciente utilizando el mismo ensayo de GH o IGF1.
> Control de la masa tumoral
La reducción o control del volumen tumoral mediante cirugía, tratamiento médico y radioterapia es un objetivo terapéutico adicional importante. Sin embargo, la medición del volumen tumoral puede ser difícil ya que se ve afectado por la cirugía previa, las diferencias en los métodos de imagen y la diferencias intraobservador.
| Prevención de las comorbilidades sistémicas |
Se están desarrollando 2 herramientas de software, SAGIT (signos y síntomas, comorbilidades asociadas, niveles de GH, IGF1 y perfil tumoral) y, ACRODAT (Herramienta de actividad de la enfermedad acormegálica), destinadas a evaluar la actividad de la enfermedad, con el objetivo de asistir a los endocrinólogos en el manejo de la acromegalia en la práctica clínica.
> Cirugía
La adenomectomía transesfenoidal sigue siendo la piedra angular del tratamiento de los tumores hipofisarios secretores de GH. Es el tratamiento de elección, excepto en aquellos pacientes con riesgo quirúrgico elevado, se nieguen a la cirugía o tienen tumores invasivos irresecables. Realizado por un cirujano experimentado, el abordaje transesfenoidal es efectivo en casi el 75% de los microadenomas y macroadenomas hipofisarios puramente intraselares.
Sin embargo, las tasas de remisión son mucho menores para los macroadenomas invasivos: 44,5% de los tumores supraselares, 33% de los tumores supraselares con compromiso visual y 41,5% de los tumores con expansión paraselar y/o esfenoidal.
Por consiguiente, aún en centros de referencia con neurocirujanos experimentados y capacitados, la curación quirúrgica no se alcanza en casi el 50% de los pacientes y es necesario hacer un tratamiento adyuvante. Hoy en día, la cirugía transcraneal rara vez es necesaria.
> Tratamientos farmacológicos
El tratamiento médico tiene un papel importante en el manejo de la acromegalia, ya sea como tratamiento de primera línea, alternativa a la cirugía o, tratamiento de segunda línea. Los ligandos del receptor de somatostatina (SRL, siglas del inglés)) de primera generación son la terapia médica de primera línea para la mayoría de los pacientes con acromegalia, aunque hay tratamientos alternativos, como los agonistas de la dopamina, el antagonista del receptor de GH pegvisomant y el SRL pasireotida de segunda generación.
Los agonistas de la dopamina tienen un papel limitado y se utilizan principalmente como tratamiento médico de primera línea en pacientes con poco aumento de los niveles de GH e IGF1, o se utilizan combinados con SRL de primera generación en pacientes que son parcialmente resistentes a los SRL.
El pegvisomant está indicado después del fallo quirúrgico o si hay resistencia al tratamiento con SRL, ya sea en monoterapia o combinado con SRL, mientras que la pasireotida está indicada cuando la cirugía no tiene éxito o no es una opción, y cuando el tratamiento con SRL de primera generación no es efectivo para controlar la acromegalia, especialmente en pacientes con tumor residual clínicamente relevante y/o alerta clínica por el crecimiento tumoral.
En casos específicos, para tratar la acromegalia se utilizan estrógenos y moduladores selectivos de los receptores de estrógenos, ya que pueden reducir los niveles de IGF1, administrados solos o en combinación con SRL o agonistas de la dopamina, aunque los pocos estudios publicados sobre el tratamiento de la acromegalia con estos agentes son insuficientes para orientar sobre su uso óptimo.
>Ligandos del receptor de somatostatina
Los SRL de primera generación se recomiendan como terapia de primera línea para los pacientes con contraindicaciones quirúrgicas o poca probabilidad de curación debido a la invasión tumoral del seno cavernoso. También se pueden utilizar como tratamiento preoperatorio, con el objetivo de mejorar la comorbilidad de la acromegalia y las complicaciones postoperatorias, y reducir el volumen tumoral, con miras a un mejor resultado quirúrgico.
Sin embargo, la Endocrine Society no los recomienda en el preoperatorio si el paciente presenta una hinchazón grave de la faringe y apnea del sueño, o insuficiencia cardíaca de alto gasto (es decir, fallo cardíaco con fracción de eyección preservada)), con el fin de reducir el riesgo quirúrgico de las comorbilidades severas.
Por el contrario, no se recomienda el uso rutinario del tratamiento médico preoperatorio para mejorar el control bioquímico posquirúrgico, ya que los resultados informados no pueden ser claramente atribuidos a la cirugía o a la terapia SRL.
Los SRL de primera generación incluyen la octreotida de liberación prolongada de acción larga o de depósito y autogel de lanreotida, considerados equivalentes en el logro del control bioquímico y clínico y en la reducción del tamaño del tumor.
La tasa de control bioquímica es ~55% para los pacientes tratados con SRL de primera generación, aunque no se han informado grandes diferencias en la eficacia hormonal de los SRL. Es difícil evaluar la eficacia global de estos fármacos debido a una importante variación en los trabajos clínicos y la metodología de los ensayos, pero oscila entre el 25% y el 70%.
Los SRL reducen el tumor hasta en el 80% de los pacientes, siendo mayor cuando se utilizan como terapia de primera línea. Sin embargo, los informes de los efectos de los SRL en el tamaño del tumor están limitados por la falta de rigor en el diseño de los estudios, la heterogeneidad de las técnicas de imagen y mediciones y la falta de controles.
Los principales predictores de la capacidad de respuesta a los SRL son el sexo (las mujeres parecen responder mejor que los hombres), la edad, los niveles basales de GH y IGF1, el volumen tumoral y, la expresión del receptor de somatostatina, en particular los subtipos SSTR2 y SSTR5.
Sobre la base del análisis histopatológico, los tumores densamente granulados son más sensibles a los SRL que los adenomas escasamente granulados, que son más comunes. Por otra parte, la presencia de un tumor hipointenso ponderado en T2, que se correlaciona con la granularidad tumoral densa, predice una respuesta favorable a los SRL.
En particular, una intensidad de señal ponderada en T2 más baja en la RM diagnóstica se correlacionó con mejor respuesta al tratamiento en términos de reducción de los niveles de GH e IGF1 y una mayor reducción del tumor después de 6 meses de tratamiento con SRL.
Aunque aún falta una definición clara de resistencia a los SRL en pacientes con acromegalia, los datos actuales indican que la capacidad de respuesta a los SRL está mejor definida cuando se tienen en cuenta los puntos finales, tanto bioquímicos como tumorales, y casi en el 25% de los pacientes tratados con un SRL durante al menos 12 meses no se logró el controlar la enfermedad.
La eficacia del tratamiento podría mejorarse aumentando la dosis de SRL o la frecuencia de las dosis, lo que mejora el control de la enfermedad en al menos el 25% más de pacientes con acromegalia.
En EE: UU. y Europa ha sido aprobada la pasireotida, un SRL de segunda generación para el tratamiento de la acromegalia. Los ensayos clínicos de fase III demostraron que la pasireotida tiene mayor efecto en el control bioquímico que la octreotida.
Los pacientes nunca tratados con SRL o que son resistentes a los SRL de primera generación Por lo tanto, PAS es una nueva opción terapéutica válida para pacientes con acromegalia, especialmente los resistente a los SRL de primera generación.
Asimismo, se produjo una reducción >25% en el volumen del tumor en más pacientes que con SRL de primera generación. Sin embargo, hasta la fecha, la pasireotida ha sido utilizada solo para el tratamiento de la acromegalia en ensayos clínicos en fase III.
Este fármaco induce hiperglucemia relacionada con efectos adversos en el 60 al 70% de los pacientes tratados, lo que puede limitar su uso en pacientes con diabetes mellitus no controlada. Por lo tanto, antes de iniciar el tratamiento con pasireotida, se debe evaluar la glucemia y conseguir su control óptimo; este monitoreo debe continuarse durante la terapia con pasireotida y cualquier alteración debe ser rápidamente corregida.
> Agonistas de la dopamina
Los agonistas de la dopamina, en particular la cabergolina, tienen un papel limitado en el tratamiento de la acromegalia y están indicados en pacientes cuya enfermedad es leve.
La monoterapia con cabergolina está indicada principalmente en los pacientes con niveles elevados de IGF1 (IGF1 2 veces el valor límite superior del rango normal ajustado por la edad) como terapia de primera línea o, como terapia de segunda línea si es efectivo en ~35% de los pacientes.
Sin embargo, la terapia combinada con SRL de primera generación está indicada en pacientes que son parcialmente resistentes a los SRL y normaliza los niveles de IGF1 en más del 50% de los pacientes.
> Pegvisomant
Según las recomendaciones de la Endocrine Society, este antagonista del receptor de GH está indicado como terapia de segunda o tercera línea, mayormente en los pacientes en los que ha fracasado la cirugía o que muestran una mala respuesta a los SRL de primera línea.
En los pacientes que responden parcialmente a los SRL, se debe considerar el tratamiento combinado de pegvisomant y SRL, mientras que el cambio a la monoterapia con pegvisomant es una estrategia terapéutica válida en pacientes con acromegalia resistente a los SRL.
En ensayos clínicos se comprobó que la monoterapia con pegvisomant normaliza los niveles de IGF1 en ~90% de los pacientes y casi el 60% de los pacientes en estudios del mundo real, probablemente debido a las diferencias en la dosificación, ya que las mayores tasas de eficacia se observan en los pacientes cuyas dosis se fueron incrementando en forma gradual. T
ambién se observa mayor eficacia cuando se usa combinado con un SRL. Los predictores de respuesta a este fármaco son edad, sexo, índice de masa corporal y niveles de GH o IGF1. En consecuencia, para normalizar los niveles de GH, los pacientes jóvenes, varones u obesos, podrían necesitar dosis más elevadas altas que otros pacientes.
| Radioterapia |
Se prefiere la radioterapia estereotáxica (preferentemente radioterapia dirigida en altas dosis) en lugar de la radioterapia convencional, a menos que haya una carga tumoral residual o el tumor se encuentre demasiado cerca del quiasma óptico.
| Calidad de vida |
Actualmente se han alcanzado resultados importantes en los resultados primarios de las enfermedades de la hipófisis, incluida la acromegalia. Se ha mejorado la tasa de mortalidad y morbilidad, la evaluación del estado funcional y la calidad de vida relacionada con la salud. Para evaluar estos parámetros se han desarrollado y validado varias herramientas, entre ellas el Acromegaly Quality of Life Questionnaire (AcroQOL).
Sin embargo, la falta de una definición inequívoca de la calidad de vida excluye la evaluación uniforme y la interpretación de la calidad de vida en los ensayos clínicos y diferentes autores pueden definir diferentes métricas y tener diferentes perspectivas.
Esto crea problemas al intentar evaluar los efectos terapéuticos en ausencia de comparaciones de estudios longitudinales entre cohortes de pacientes con enfermedad activa y pacientes en remisión, o cohortes sometidas a diferentes modalidades de tratamiento. Otra herramienta útil es el Leiden Bother and Needs Questionnaire for patients with pituitary disease (LBNQ- Pituitary).
> Calidad de vida en pacientes con acromegalia
La calidad de vida en pacientes con acromegalia se reduce, independientemente de la actividad de la enfermedad. Los factores más frecuentes y consistentes que influyen negativamente en la calidad de vida en los pacientes con acromegalia son: radioterapia previa, osteoartritis, afecciones articulares, dolor musculoesquelético, entumecimiento de los dedos, hipertensión, disfunción sexual, síntomas depresivos y comorbilidades de la edad avanzada actual y comorbilidades persistentes.
Sin embargo, aún se desconoce cuál es el aporte de diferentes determinantes de la calidad de vida durante las diferentes etapas de la acromegalia.
En los pacientes con acromegalia están afectados casi todos los sistemas orgánicos debido al exceso de GH e IGF1, incluyendo el cerebro. En consecuencia, además de los síntomas clínicos se han descrito cambios en la personalidad y el comportamiento, como rasgos de personalidad inadaptada y psicopatológicos.
Aunque muchos síntomas mejoran después del tratamiento, el 77% de los pacientes en remisión continuó informado problemas articulares. Se destaca que la presencia de los síntomas articulares no estuvo relacionada con las concentraciones de GH e IGF1 ni con la actividad, duración de la enfermedad o la edad.
Los pacientes experimentan la alteración de su autoimagen y apariencia, independientemente del estado de la enfermedad, lo que también afecta negativamente la calidad de vida. Una revisión sistemática de los predictores de calidad de vida halló que los efectos de las medidas de control bioquímico sobre la calidad de vida fueron mixtos y no concluyentes.
Luego del análisis de diversos estudios y metaanálisis, los autores sostienen que lograr el control bioquímico per se o el tratamiento general no es suficiente para normalizar la calidad de vida. Se requieren estudios que investiguen los predictores de calidad de vida en pacientes sin tratamiento previo, y su seguimiento después de haber recibido una intervención terapéutica.
La consistente asociación de algunos factores como la falta de percepción de la enfermedad, la depresión y la obesidad con una peor calidad de vida es un ejemplo de que las estrategias terapéuticas en la acromegalia no deben centrarse únicamente en la normalización de los marcadores bioquímicos.
Para mejorar la calidad de vida de los pacientes con acromegalia, las estrategias de tratamiento también incluyen las medición del resultado orientadas al paciente, como la atención de las necesidades del paciente (por ej., apoyo, intervenciones para bajar de peso), lo que probablemente mejore los efectos negativos o adversos de la enfermedad y las intervenciones terapéuticas.
| Perspectivas |
Los criterios de curación han evolucionado hasta los criterios actuales, con el objetivo de controlar estrictamente la enfermedad, reduciendo la mortalidad y la morbilidad y mejorando la calidad de vida.
Se recomienda el estrecho monitoreo de los pacientes que están recibiendo terapias el control de la enfermedad. El tratamiento óptimo debe ser personalizado, teniendo en cuenta la edad, las comorbilidades, el tamaño e invasividad del tumor y los niveles de GH e IGF1.
El tratamiento de la acromegalia ha cambiado con el tiempo debido a la introducción de nuevas terapias, aunque en la mayoría de los casos, el tratamiento de elección sigue siendo la cirugía hipofisaria.
Sin embargo, la introducción y el desarrollo de nuevas terapias deben mejorar el control de la enfermedad. Las nuevas terapias medicas que actualmente se encuentran en desarrollo clínico son las nuevas formulaciones de los SRL, como las cápsulas de octreotida oral, la octreotida parenteral y un nuevo oligonucleótido antisentido dirigido al receptor de GH.
Otras terapias prometedoras están en las primeras etapas de desarrollo, siendo necesaria la realización de ensayos de fase III para establecer la eficacia y seguridad de estos medicamentos y su papel en el manejo óptimo de la acromegalia. A pesar de estos logros en el tratamiento médico y la disponibilidad de nuevas técnicas de radioterapia, los adenomas hipofisarios secretores de GH agresivos y resistentes siguen siendo una preocupación.
Se están desarrollando nuevas terapias para mejorar la tasa de respuesta global. A pesar de que los beneficios del control de la enfermedad están bien establecidos, existe una clara necesidad de evaluar completamente los costos del tratamiento, incluidos los beneficios económicos potenciales de la terapia médica de por vida y su efecto sobre la calidad de vida de estos pacientes.
Por otra parte, se necesita más investigación para abordar muchas preguntas que quedan sobre la patogénesis de los tumores esporádicos secretores de GH y para comprender mejor los mecanismos de la generación de tumores hipofisarios, lo que debería facilitar el desarrollo de marcadores de agresividad tumoral así como nuevas terapias dirigidas.
Resumen y comentario objetivo: Dra. Marta Papponetti
Contenidos relacionados
Los editores le recomiendan continuar con las siguientes lecturas:




















.png)

.png)







No hay comentarios:
Publicar un comentario