Una revisión y puesta al día | 12 MAY 19
Mieloma múltiple
Resumen de los conceptos vertidos en la 3ª Clase Magistral 2019 del Royal College of Physicians sobre diagnóstico y tratamiento del mieloma múltiple

Autor: Royal College of Physicians,a la orden del Dr John Firth. Clinical medicine. 2019. Vol 19. Nº1. 57-60.
| Etiología/fisiopatología |
El mieloma múltiple (MM) se caracteriza por la acumulación de células plasmáticas clonales malignas en la médula ósea.La causa es desconocida; en algunos casos, puede estar provocado a la radiación (aunque no hay asociación con la radiación terapéutica), la exposición a toxinas industriales o agrícolas, y también se ha propuesto una asociación con los virus pero faltan pruebas.
Se han identificado anomalías cromosómicas, más comúnmente involucrando al interruptor de la región de cadena pesada de inmunoglobulinas (Ig) (en el brazo largo del cromosoma 14), aunque esto. por sí solo, no parece ser suficiente para dar lugar al MM).
Las células tumorales en la médula ósea están soportadas por una población de células estromales no malignas que producen citocinas (p. ej., interleucina-6), cuya acción potencia el crecimiento celular del mieloma y previene la apoptosis.
| Epidemiología |
La edad media al momento del diagnóstico es de 65 años. La presentación en menores de 40 años ocurre en menos del 3% de los pacientes.
| Presentación clínica |
• Dolor óseo y fracturas patológicas.
• Anemia (insuficiencia de la médula ósea)
• Infecciones recurrentes (por inmunoparesia)
• Hipercalcemia
• Insuficiencia renal (etiologías múltiples: hipercalcemia, deposición de cadenas livianas, medicamentos no esteroideos, anemia, infecciones)
• Sangrado anormal (debido a disfunción plaquetaria)
> Rara
• Síndrome de hiperviscosidad (isquemia, insuficiencia cardíaca y problemas neurológicos)
• Enfermedad amiloide (por ej., síndrome del túnel carpiano)
| Investigaciones |
• Hemograma completo: puede revelar anemia normocrómica, normocítica.
• Velocidad de sedimentación elevada (la carga positiva de proteína neutraliza la carga negativa del ácido siálico en la membrana eritrocítica, reduciendo su tendencia natural a rechazarse entre sí y haciendo que las células se depositen más rápido en una columna).
• Determinación de la hipercalcemia (generalmente con una fosfatasa alcalina normal) y alteración renal.
• Electroforesis sérica: puede demostrar una proteína monoclonal, que en la mayoría de los casos es IgG o IgA, pero puede ser cualquier clase de Ig. Los niveles reducidos de Ig normales son confirmatorios.
• En el suero puede hallarse el exceso de cadenas livianas libres, ya sea kappa o lambda (a veces el clon de células plasmáticas es solo de cadenas livianas).
• El diagnóstico de MM depende de la demostración del aumento de células plasmáticas (>10%) en la médula ósea.
• Las radiografías óseas pueden mostrar lesiones líticas.
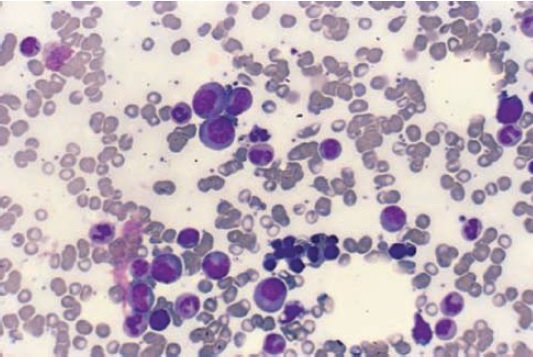
Células de mieloma. Esta médula ósea muestra diferentes tipos de células. Las células más grandes con núcleos excéntricos y citoplasma basófilo son células de mieloma. Tenga en cuenta la transparencia perinuclear que representa el aparato de Golgi.
| Estadificación (Sistema Internacional de Estadificación) |
| Signos del mieloma | ||
| Estadio 1 | Estadio 2 | Estadio 3 |
| ß2-Ig < 3,5 mg/dl | Ni 1 ni 3 | ß2-Ig >5,5 mg/dl |
| Albúmina >35 g/l | ||
| Diagnóstico diferencial de una paraproteína sérica |
• Macroglobulinemia de Waldenström
• Linfoma
• Leucemia linfocítica crónica
• Amiloidosis primaria
• Leucemia de células plasmáticas
Benigno/estable
• Gammapatía monoclonal de significado incierto.
• SIDA
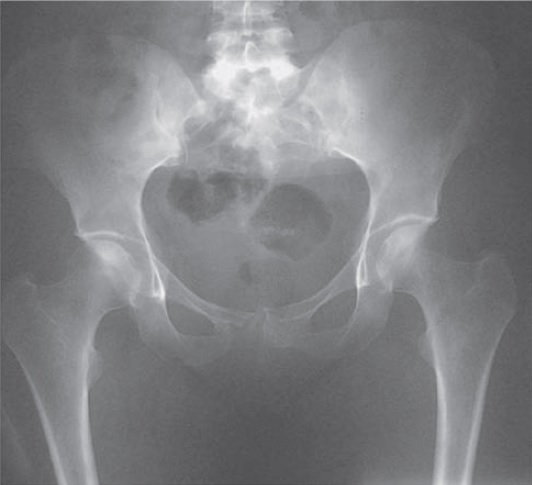
Mieloma de hueso. Tenga en cuenta que la sínfisis del pubis ha sido erosionada por el mieloma. No hay depósitos aparentes en los fémures superiores o pelvis.
| Tratamiento |
• Lesión renal aguda: es muy importante hacer el tratamiento inmediato de la depleción de volumen, consultar tempranamente al nefrólogo.
• Hipercalcemia: son importantes los líquidos y los bifosfonatos.
• Compresión de la médula espinal: radioterapia de emergencia.
• Hiperviscosidad: considerar la plasmaféresis.
El tratamiento específico depende de una serie de factores como las comorbilidades y la presencia de factores de mal pronóstico. No hay cura conocida para el MM, pero en los últimos años los tratamientos han mejorado en gran medida. Ahora, muchos pacientes pueden lograr una remisión estable, que dura varios años, a través de una combinación de quimioterapia y trasplante de células madre autólogas.
Muchos hematólogos participarán en ensayos asegurando estándares terapéuticos y acceso a medicamentos que solo pueden estar disponibles para los pacientes que participan en esos ensayos. (es decir, están financiados para los pacientes participantes).
> Quimioterapia
La quimioterapia combinada con esteroides, es la base del tratamiento para los pacientes recién diagnosticados. En los últimos 10-15 años se han desarrollado nuevos tratamientos, incluyendo la talidomida y la lenalidomida (cuyo mecanismo de acción todavía está bajo investigación pero que posiblemente sea antiangiogénico) y el bortezomib (un inhibidor del proteasoma).
Las toxicidades de estos fármacos incluyen eventos tromboembólicos y neuropatía periférica. Estos regímenes se pueden administrar en pulsos (es decir, repetirlos a intervalos de tiempo regulares), ya sea por vía oral o subcutánea.
Las tasas de respuesta son elevadas: algunos pacientes entran en remisión completa, pero la mayoría permanece en una fase de meseta en la que la la paraproteína es más baja pero constante.
Eventualmente, todos los pacientes recaen. Se utilizan regímenes más intensivos en los pacientes más jóvenes y físicamente aptos; estos medicamentos pueden ser usados para reducir la enfermedad antes del trasplante.
> Trasplante autólogo de células madre
La seguridad actual de estos procedimientos ha permitido su aplicación a un mayor número de pacientes. Muchos hematólogos considerarían pacientes de hasta 70 años como elegibles para trasplantes autólogos, como parte de su tratamiento de primera línea (es decir, reducir primero con quimioterapia y luego proceder directamente al trasplante).
Esto depende de la condición general del paciente y de su respuesta a la quimioterapia. También se puede realizar un segundo trasplante, a menudo con buenos resultados.
> Trasplante alogénico de células madre
Esta opción es solo para una minoría de pacientes, particularmente aquellos que son jóvenes, físicamente aptos y tienen un hermano donante de antígeno leucocitario humano compatible (o bien el donante es compatible y no relacionado). Las tasas de respuesta son elevadas, pero lamentablemente la recaída es común.
> Terapia de mantenimiento
La lenalidomida se usa como terapia de mantenimiento después de la recaída, para ayudar a prolongar la remisión. Las toxicidades son el tromboembolismo venoso (por lo que a menudo, se trata de prevenirlo con la anticoagulación), calambres y citopenias.
> Plasmaféresis
La viscosidad del plasma puede proporcionar una indicación para la plasmaféresis pero mucho más importantes son las características clínicas. La isquemia grave, el síndrome neurológico o el coma pueden mejorar después de esta intervenciónn
> Cuidados de apoyo
La hipercalcemia debe tratarse inicialmente con líquidos y b bifosfonatos. Los bifosfonatos deben continuarse mensualmente, incluso con un nivel de calcio normal, ya que se ha propuesto que pueden reducir la enfermedad ósea y modular el trastorno.
El dolor es común en el MM y con frecuencia requiere analgesia con opiáceos, además de los antiinflamatorios no esteroides. La radioterapia puede ser útil para controlar el dolor provocado por las lesiones óseas localizadas.
Pueden requerirse transfusiones de sangre y antibióticos en forma rrepetida, como resultado tanto de la enfermedad como del tratamiento. En los pacientes que tienen infecciones recurrentes, se pueden considerar las infusiones profilácticas de Ig por vía intravenosa.
> Apoyo psicológico
Casi con seguridad, el paciente necesitará ayuda al recibir el diagnóstico. Trate de involucrar a la familia o los amigos, particularmente para que lo acompañe en ese momento. Se debe aconsejar al paciente que se conecte con instituciones o grupos de apoyo donde pueda recibir este tipo de ayuda.
> Pronóstico
La supervivencia media con quimioterapia es de aproximadamente 3 a 5 años. Un estadio elevado se correlaciona con mal pronóstico.
Resumen y comentario objetivo: Dra. Marta Papponetti
Contenidos relacionados
Los editores le recomiendan continuar con las siguientes lecturas:






















.png)








No hay comentarios:
Publicar un comentario