Logran las primeras simulaciones estables de cristales de ADN
Un equipo del Instituto de Investigación Biomédica de Barcelona ha conseguido por primera vez simulaciones estables de cristales de ADN. El estudio presenta la descripción más detallada a nivel atómico de las propiedades de sistemas cristalinos con ADN presentada hasta la fecha.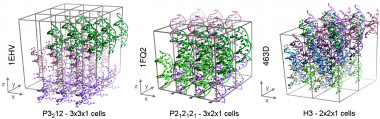
Representación de los tres sistemas cristalinos estudiados en este trabajo, que contienen respectivamente (de izquierda a derecha) 27, 24 y 36 moléculas doble-hebra de ADN. / Pablo Dans Puiggròs / IRB Barcelona
La cristalografía de rayos-X ha sido la técnica más usada desde el nacimiento de la biología estructural para determinar la estructura en tres dimensiones de las biomoléculas, los compuestos químicos que se encuentran en los organismos vivos. Esta técnica podría mejorarse si se conocieran las interacciones entre las biomoléculas con su entorno cristalino y las fuerzas moleculares que estabilizan los cristales.
Un estudio publicado en la revista Chem, del grupo Cell, y elaborado por investigadores del Instituto de Investigación Biomédica (IRB Barcelona), ha logrado por primera vez simulaciones estables de cristales de ADN. Este hito ha permitido explicar la importancia de los aditivos químicos que se usan experimentalmente para lograr las condiciones de cristalización y obtener cristales estables en el laboratorio.
Biofísicos y fisicoquímicos computacionales cuentan ahora con una referencia y protocolos claros para obtener simulaciones estables
“Los beneficiarios de estudio son los biofísicos y fisicoquímicos computacionales, quiénes cuentan ahora con una referencia y protocolos claros para obtener simulaciones estables de cristales de ADN”, afirma Pablo D. Dans, investigador postdoctoral del IRB Barcelona.
El estudio liderado por Modesto Orozco, jefe del laboratorio de Modelización Molecular y Bioinformática, presenta la descripción más detallada a nivel atómico de las propiedades de sistemas cristalinos con ADN presentada hasta la fecha.
Predecir el efecto de un aditivo químico
“A largo plazo, la simulación de diversos cristales obtenidos en diferentes condiciones experimentales debería permitir anticipar y predecir el efecto de un aditivo químico dado, guiando a los cristalógrafos en sus experimentos y reduciendo considerablemente los costos y los tiempos de obtención de cristales”, subraya Modesto Orozco, cuyo laboratorio es uno de los referentes mundiales en computación y simulación de biomoléculas.
El estudio ha recibido fondos del Consejo Europeo de Investigación (ERC), a través del proyecto ERC Advanced Grant (SimDNA) otorgado a Modesto Orozco, y del Consorcio Europeo de Centros de Supercomputación a través de la beca PRACE 12th Call, otorgada a los tres autores del estudio.
Referencia bibliográfica:
Antonija Kuzmanic, Pablo D. Dans and Modesto Orozco. “An in-depth look at DNA crystals through the prism of molecular dynamics simulations”. Chem (2019) DOI: 10.1016/j.chempr.2018.12.007























.png)








No hay comentarios:
Publicar un comentario