Potential inhibitors of SARS-CoV-2 identified

In the absence of an effective drug or vaccine, the fight against severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), the agent which causes COVID-19, has been slow and ineffectual. Now, a new study by researchers at the University of Texas and published on the preprint server bioRxiv* in August 2020 reports the discovery of two inhibitors of the helicase enzyme, which is essential for viral replication and the most highly conserved non-structural coronavirus (CoV) protein.
Atomic Structure of Viral Protein Nsp13
The study focuses on clarifying the atomic structure of the viral non-structural protein SARS-CoV2 Nsp13. While there are some structurally homologous structures in published literature, none of them are useful for the analysis of molecular docking. There are apo-nsp13 crystal structures for the SARS- and MERS-CoV, both of which show two identical chains in their crystal lattice. These are S1A and S1B, and M1A and M1B, respectively.
The two viral proteins differ mainly in one loop of the Rec1A Rec1A10 domain, which interacts with domain 1B. The M1A does not have this because of its highly dynamic nature. There are various other differences between nsp13 structures in different viruses, and owing to the high flexibility of the structure, it is important to have multiple templates or models showing the helicase in many different conformations since it is highly flexible. Any of these might prove superior to the rest in providing a substrate for high-affinity inhibitors.
To help achieve this, the researchers created an array of inhibitors with affinity to SARS and SARS-CoV-2 nsp13 homology models in either the apo state or the substrate-bound state. They carried out docking analyses in silico and did high-throughput virtual screening (HTvS). Using multiple analytic models, they looked for potential inhibitor compounds.
Using SARS-CoV Nsp13 to Model the SARS-CoV2 Nsp13
Since the helicase in both viruses is 99.8% identical in sequence, the researchers solved the apo-SARS-CoV nsp13 helicase crystal structure. They found two chains in the unit of this structure, indicating that it is inherently flexible. They consider this an ideal screening structure since there is only one amino acid difference between the two proteins. On the other hand, the researchers found that there were issues with the quality of the published data, and a poorly fitted loop with improbable sequences.
A more serious error was the lack of the typical sulfate ions that were not found to be bound even without the obvious requirement for these ions. The researchers, therefore, regenerated the model on the basis of the MERS nsp13, which is 72% similar to the SARS-CoV-2 nsp13, correcting existing flaws to create a new crystal structure. They rebuilt the Walker-A loops and sulfate ions in both chains using the apo crystal structure.
Using various techniques, they finally came up with two very similar energy-minimized models for the two viral chain pairs, without stereochemical flaws. They observed only a few differences between them in the 1B domain, zinc-binding, and ATP-clamping Rec2A domains. They report that their model of the M2B chain is “the only complete apo apo-Nsp13 structure without gaps in the model.”
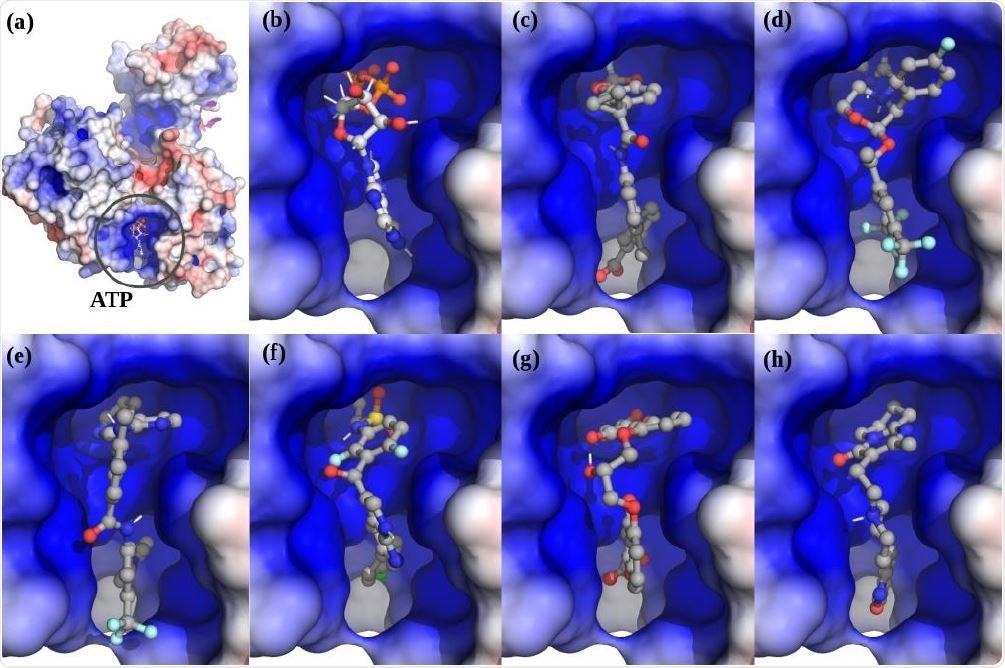
. The top scoring hits for the Nsp13 complex ATP ATP-site using virtual screening. (a) The Nsp13:ATP:RNA complex. (b) ATP. (c) Lumacaftor. (d) Emend (Aprepitant). Row Row-2: (e) Nilotinib. (f) Zelboraf. (g) Cromolyn. (h) Risperdal.
Building an ATP- or RNA-Bound SARS-CoV-2 nsp13 Model
In order to build a model of the SARS-CoV-2 helicase in complex with ATP or ssRNA, the researchers used the yeast Upf1 helicase complex13 because of its high structural homology to the MERS/SARS nsp13 helicases. This allows the domains to be aligned by secondary structure, providing a foundation for the creation of the enzyme: ATP or RNA complexed structure before minimizing the energy of the final structure.
They achieved a good fit, and then performed in silico HTvS on all the five homology models. This showed 369 selected drugs, which were assigned to 93 exemplar compounds by chemical affinity propagation clustering, illustrating how diverse the selected drugs are. They then set a limit cut-off to filter them to a lower number of inhibitors used for in vitro or in vivo screening. The use of multiple models allowed overlapping and non-overlapping hits to be identified, including one ATP-binding site and four apo ATP sites, the latter being unbound.
Top-Scoring Drugs
The four apo-ATP sites showed several possible helicase targets, with the top contenders being cepharanthine, cefoperazone, dihydroergotamine, and cefpiramide. Among those molecules that docked with the ATP-binding site, the top-scoring compounds were lumacaftor, emend, nilotinib, and irinotecan. Despite the different conformations at the ATP-binding pocket, the top 20 drugs in both models include lumacaftor, nilotinib, liftegrast, idarubicin, and irinotecan.
Nilotinib and lumacaftor were the top-scoring docking molecules for the ATP site in both models. Nilotinib is a BCR-ABL tyrosine kinase inhibitor, used to treat chronic myelogenous leukemia. It was designed on the foundation of the crystal structure of the complex formed by imatinib and ABL protein, in order to dock at the ATP-binding site of the protein with increased affinity. It may also inhibit the nsp12-nsp7-nsp8 complex of SARS-CoV-2, which mediates RNA‐dependent RNA polymerase activity. The latter is essential for RNA synthesis and hence for viral replication.
Lumacaftor supervises protein folding and enhances the processing of F508del, the most common of the CFTR mutants, responsible for cystic fibrosis, and its transport to the cell surface. It is also part of the treatment regimen for this condition, associated with this deletion mutant.
Cepharanthine was the top-scoring hit for the apo-ATP site on screening, and lumacaftor for complex-ATP site screening. The former is known to be an inhibitor of SARS, and of SARS-CoV-2, via high-throughput drug screens and in siRNA assays. It also suppresses the activity of the SARS-CoV-2 Nsp12-Nsp8-Nsp7 complex. The current study shows its potential for synergistic action on nsp13 within the viral replication complex. It also provides an incentive to study the inhibitory activity of lumacaftor in cell-based and animal-based models.
Implications
The researchers sum up: “We have performed extensive integrated structural modeling to build atomic structural models of SARS-CoV2 Nsp13 in its apo-and substrate-bound conformations. Two of our top HTvS hits show significant activity in inhibiting purified recombinant SARS-CoV-2 helicase.” The study, therefore, indicates the potential for re-purposing these drugs for COVID-19 therapy.
Conclusion
In conclusion, virtual molecular docking analyses targeting the ATP binding pocket using these structural models have led to the identification of potential inhibitor compounds, many of them approved human drugs. Of particular interest, providing hope that these drugs can be potentially re-purposed for the treatment of COVID
*Important Notice
bioRxiv publishes preliminary scientific reports that are not peer-reviewed and, therefore, should not be regarded as conclusive, guide clinical practice/health-related behavior, or treated as established information.
Journal reference:
- White, M. A. et al. (2020). Discovery of COVID-19 Inhibitors Targeting the SARS-CoV2 Nsp13 Helicase. bioRxiv preprint. doi: https://www.biorxiv.org/content/10.1101/2020.08.09.243246v1






















.png)








No hay comentarios:
Publicar un comentario